Briefly explain the results you obtained. You can use figures to illustrate the results, however, figures must be a readable size and of publication quality resolution. It is essential that you explain what you find; i.e. you need to explain the results using your own words. Figures and the outputs of the analysis software alone will only earn partial marks: the most important aspect is to demonstrate your understanding of the result.
Provide your code with proper annotations.
The answers for Tasks 1-3, including figures and scripts, must not exceed 2 pages for each task and for task 4 must not exceed 300 words. Please use font size 10 and double spacing.
1. Download any RNAseq datasets for an experiment. You can use your own data or find one of interest from NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/ ). You must identify a dataset comparing at least two conditions (you may use the example shown in class), and align these to the appropriate reference genome. Perform basic data quality assessment using FastQC or other software, trim data if necessary. Please don’t be over-ambitious and discuss with the lecturer about the dataset you selected if necessary.
The answers for Tasks 1-3, including figures and scripts, must not exceed 2 pages (Please use font size 10 and double spacing)
---
The RNAseq datasets used in this experiments were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128699. The datasets were obtained from single-nucleotide resolution m6A mapping (miCLIP) on wild-type and METTL5 knockout HCT116 cells to identify the METTL5-dependent methylation events.
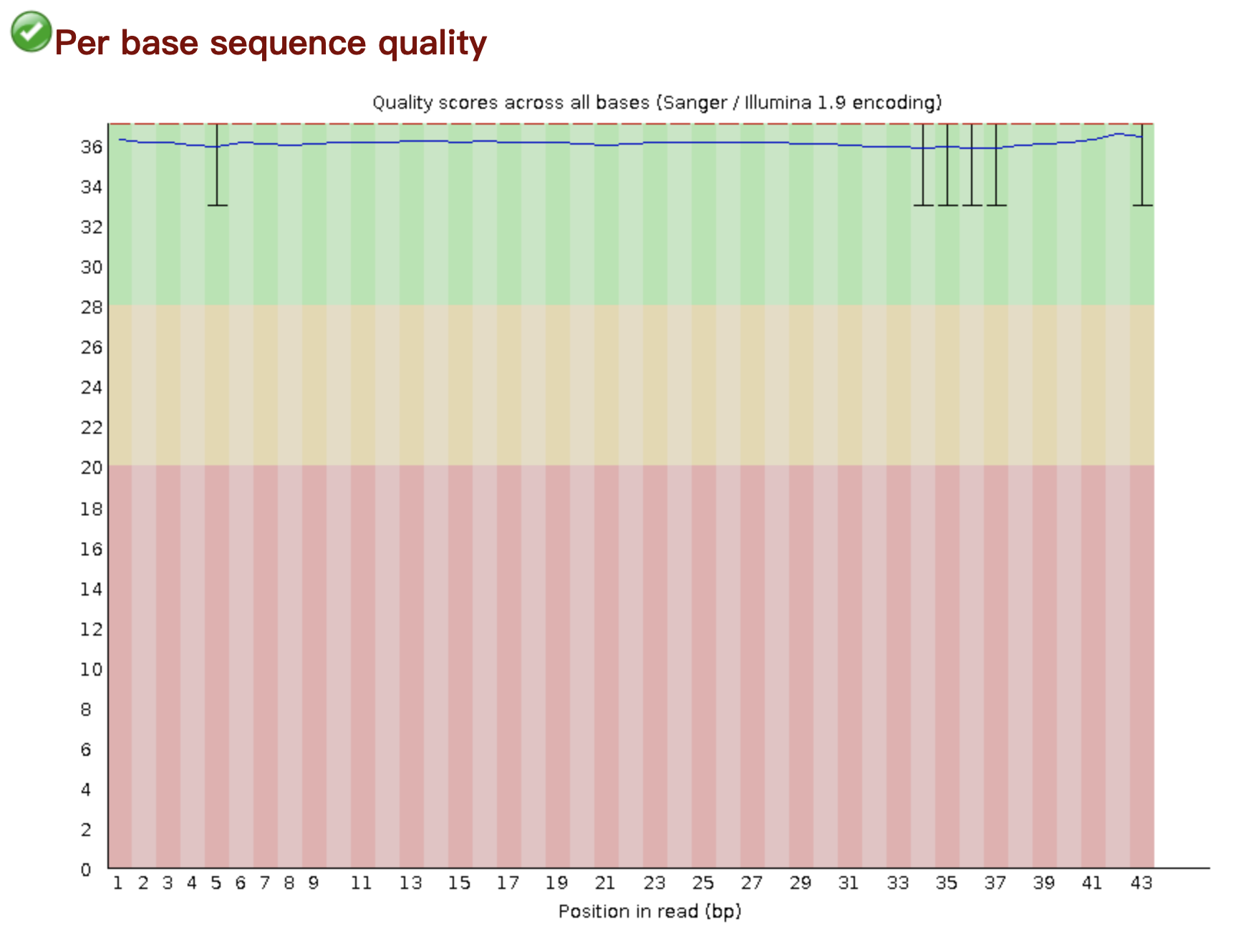
Once the datasets were successfully downloaded to the server, quality control method would be used by "fastqc". In this case the sequence data were paired-end, which required me to check the data quality of both ends.
1 2 3 4 5 6
# S4. quality control #################### # for paired end of data, you have to check the data quality of both reads fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767363_1.fastq $dir_fastq/SRR8767363_2.fastq fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767364_1.fastq $dir_fastq/SRR8767364_2.fastq fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767365_1.fastq $dir_fastq/SRR8767365_2.fastq fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767366_1.fastq $dir_fastq/SRR8767366_2.fastq
All the data achieved high performance, but considering the page limits, I would randomly select one sample to illustrate the results.
wget -O ~/cw1/P1/index/hg38.fa http://10.7.88.74/MyWeb/Genomes/hg38/genome.fa # download the genome wget -O ~/cw1/P1/index/hg38.gtf http://10.7.88.74/MyWeb/Genomes/hg38/genes.gtf # download the annotation
# S6 build index, needed for alignment (nohup hisat2-build ~/cw1/P1/index/hg38.fa ~/cw1/P1/index/hg38 > ~/cw1/P2/index/build_index.out)& # build index, take a very long time #the index is necessary for the alignment and increase the query rate
# S10 index all the bam files, which is required for visualization in IGV (nohup samtools index ~/cw1/P1/bam/SRR8767363_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/cw1/P1/bam/SRR8767364_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/cw1/P1/bam/SRR8767365_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/cw1/P1/bam/SRR8767366_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file.
# S11 visualization of bam files wget -O ~/cw1/P1/tdf/hg38.chrom.sizes http://10.7.88.74/MyWeb/Genomes/chrom.sizes/hg38.chrom.sizes # use a different genome size file if necessary
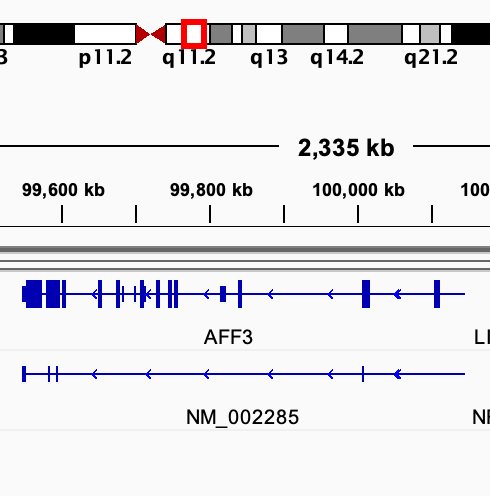
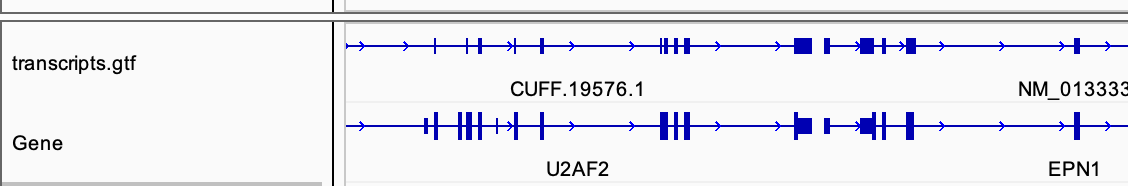
# use IGV to visualize the TDF files # use IGV to visualize the BAM files (with the .bai index file) # compare the tdf file generated with the gtf files
# S1. make your working directory P1 ################################## dir="cw1" cd$dir
P1="P1" if [ ! -d $P1 ]; then mkdir $P1 else echo"the file of $P1 exists!" fi
# S2. download the raw data in sra format #################################### # Data from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128699
# specify the directory inside P1 ~cw1/P1/fastq
dir_p1=$(pwd)/P1 fastq="fastq" if [ ! -d "$dir_p1/$fastq" ]; then mkdir "$dir_p1/$fastq" else echo"the file of $fastq exists!" fi
# ZCCHC4 knockout (no use in this case) (nohup fastq-dump --split-3 SRR8767367 -O $dir_fastq > $dir_fastq/230.out)& (nohup fastq-dump --split-3 SRR8767368 -O $dir_fastq > $dir_fastq/231.out)&
# S4. quality control #################################### fastqc_report="fastqc_report" dir_fastqc_report="$dir_p1/$fastqc_report" if [ ! -d $dir_fastqc_report ]; then mkdir $dir_fastqc_report else echo"the file of $fastqc_report exists!" fi
fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767363_1.fastq $dir_fastq/SRR8767363_2.fastq fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767364_1.fastq $dir_fastq/SRR8767364_2.fastq fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767365_1.fastq $dir_fastq/SRR8767365_2.fastq fastqc -o $dir_fastqc_report -f fastq $dir_fastq/SRR8767366_1.fastq $dir_fastq/SRR8767366_2.fastq # for paired end of data, you have to check the data quality of both reads
# S5 obtain the reference genome
wget -O ~/cw1/P1/index/hg38.fa http://10.7.88.74/MyWeb/Genomes/hg38/genome.fa # download the genome wget -O ~/cw1/P1/index/hg38.gtf http://10.7.88.74/MyWeb/Genomes/hg38/genes.gtf # download the annotation
# S6 build index, needed for alignment (nohup hisat2-build ~/cw1/P1/index/hg38.fa ~/cw1/P1/index/hg38 > ~/cw1/P2/index/build_index.out)& # build index, take a very long time #the index is necessary for the alignment and increase the query rate
# S10 index all the bam files, which is required for visualization in IGV (nohup samtools index ~/cw1/P1/bam/SRR8767363_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/cw1/P1/bam/SRR8767364_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/cw1/P1/bam/SRR8767365_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/cw1/P1/bam/SRR8767366_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file.
# S11 visualization of bam files wget -O ~/cw1/P1/tdf/hg38.chrom.sizes http://10.7.88.74/MyWeb/Genomes/chrom.sizes/hg38.chrom.sizes # use a different genome size file if necessary
# use IGV to visualize the TDF files # use IGV to visualize the BAM files (with the .bai index file) # compare the tdf file generated with the gtf files
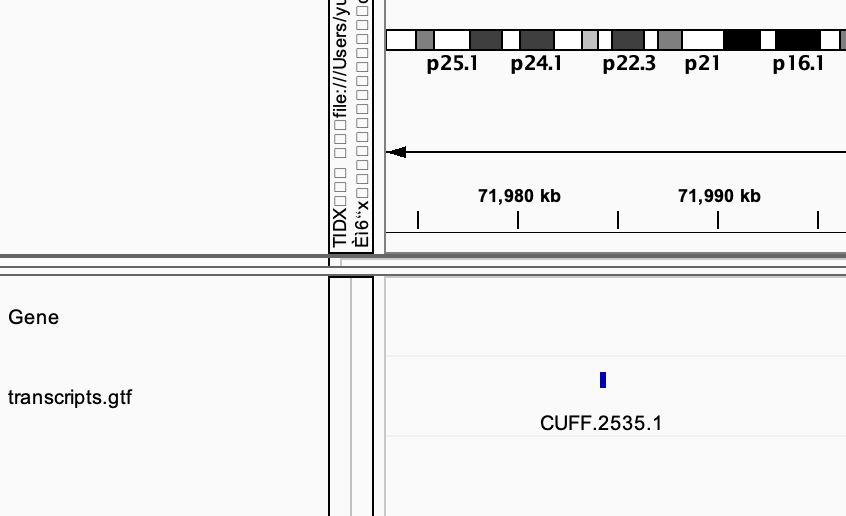
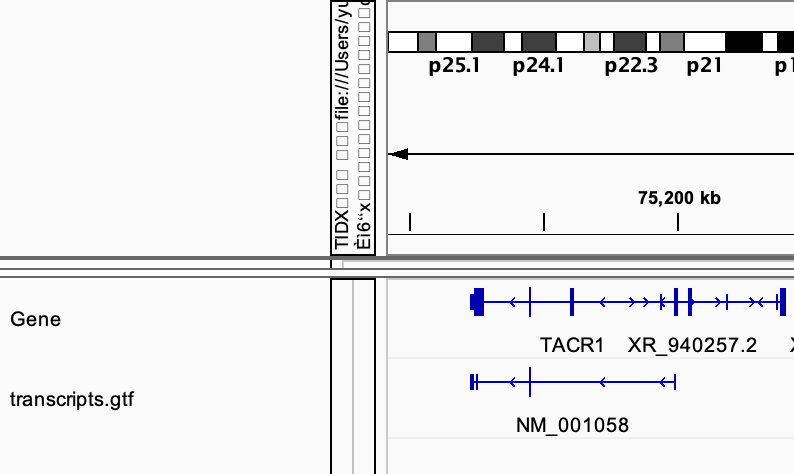
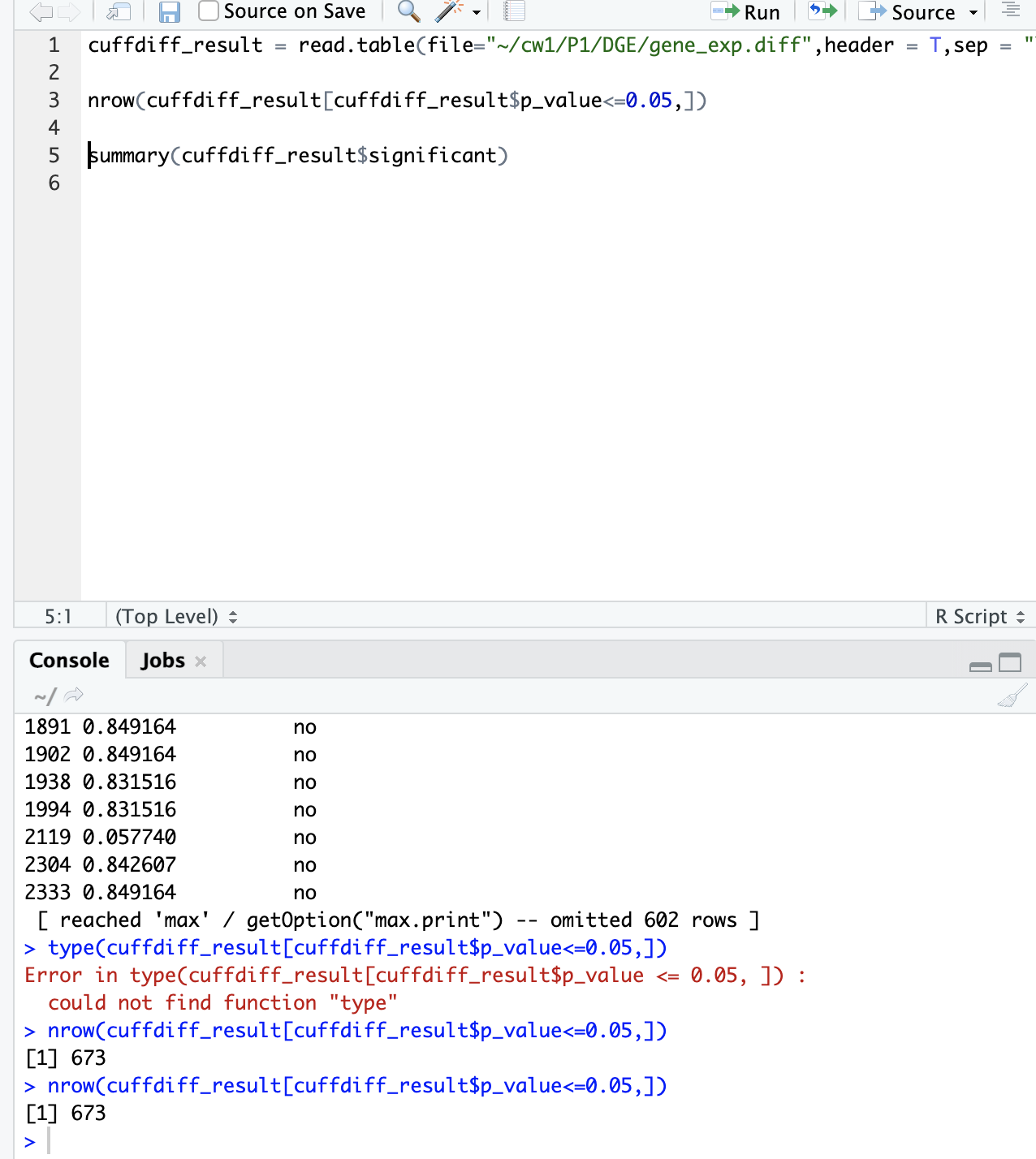
cufflinks --help# check how to do reference-based transcriptome assembly # view the gtf file in IGV # compare it with the standard gtf file provided in IGV