RNA-seq (RNA-sequencing) is a technique that can examine the quantity and sequences of RNA in a sample using next generation sequencing (NGS). It analyzes the transcriptome of gene expression patterns encoded within our RNA.
What are the applications of RNA-seq?
RNA-seq lets us investigate and discover the transcriptome, the total cellular content of RNAs including mRNA, rRNA and tRNA. Understanding the transcriptome is key if we are to connect the information on our genome with its functional protein expression.
RNA-seq can tell us which genes are turned on in a cell, what their level of expression is, and at what times they are activated or shut off. This allows scientists to more deeply understand the biology of a cell and assess changes that may indicate disease. Some of the most popular techniques that use RNA-seq are transcriptional profiling, SNP identification, RNA editing and differential gene expression analysis.
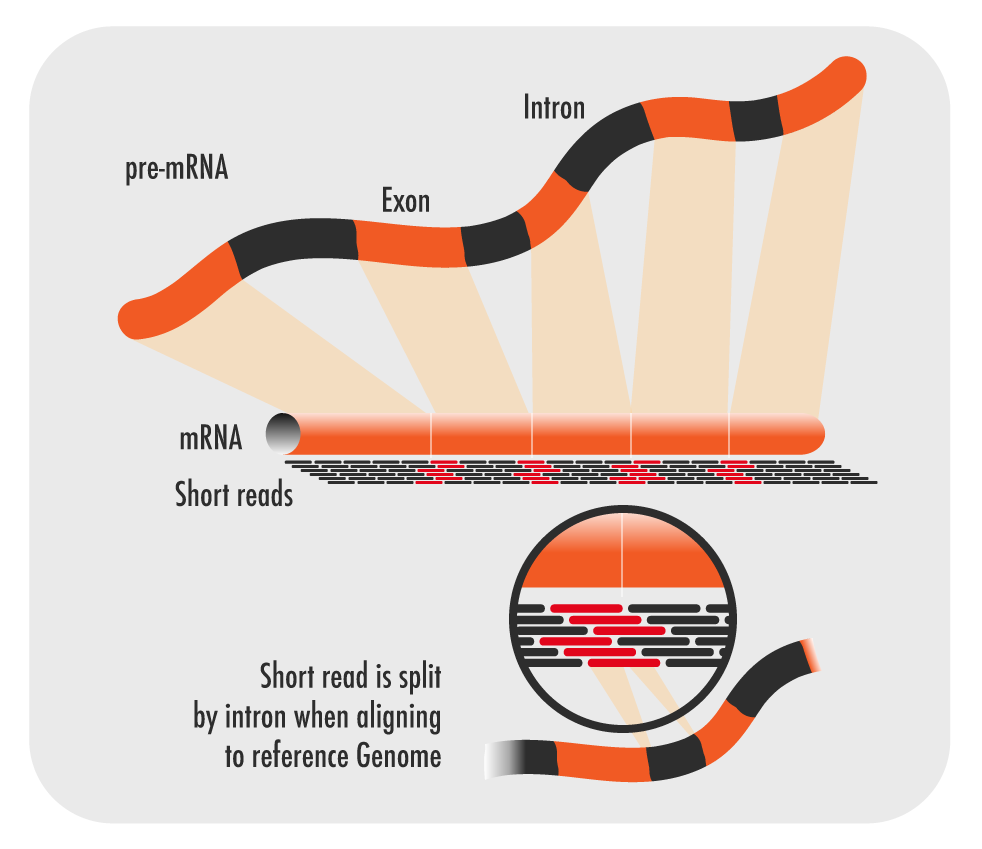
Figure1: RNA-seq data uses uses short reads of mRNA which is free of intronic non-coding DNA. These reads must then be aligned back to the reference genome.
This can give researchers vital information about the function of genes. For example, the transcriptome can highlight all the tissues in which a gene of unknown function is expressed, which might indicate what its role is. It also captures information about alternative splicing events (Figure 1), which produce different transcripts from one single gene sequence. These events would not be picked up by DNA sequencing. It can also identify post-transcriptional modifications that occur during mRNA processing such as polyadenylation and 5’ capping
How does RNA-seq work?
Early RNA-seq techniques used Sanger sequencing technology, a technique that although innovative at the time, was also low-throughput, costly, and inaccurate. It is only recently, with the advent and proliferation of NGS technology, have we been able to fully take advantage of RNA-seq’s potential
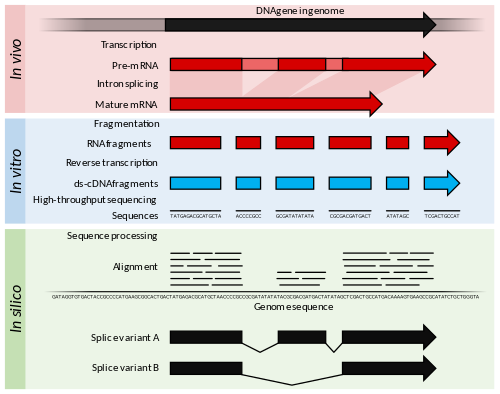
The first step in the technique involves converting the population of RNA to be sequenced into cDNA fragments (a cDNA library). This allows the RNA to be put into an NGS workflow. Adapters are then added to each end of the fragments. These adapters contain functional elements which permit sequencing; for example, the amplification element and the primary sequencing site. The cDNA library is then analyzed by NGS, producing short sequences which correspond to either one or both ends of the fragment. The depth to which the library is sequenced varies depending on techniques which the output data will be used for. The sequencing often follows either single-read or paired-end sequencing methods. Single-read sequencing is a cheaper and faster technique (for reference, about 1% of the cost of Sanger sequencing) that sequences the cDNA from just one end, whilst paired-end methods sequence from both ends, and are therefore more expensive and time-consuming5,6.
A further choice must be made between strand-specific and non-strand-specific protocols. The former method means the information about which DNA strand was transcribed is retained. The value of extra information obtained from strand-specific protocols make them the favorable option.
These reads, of which there will be many millions by the end of the workflow, can then be aligned to a genome of reference and assembled to produce an RNA sequence map that spans the transcriptome7.
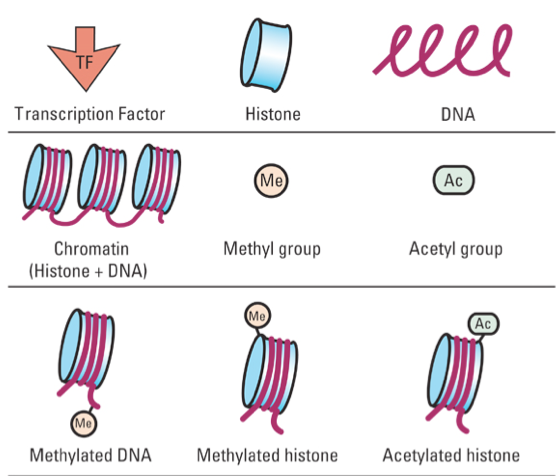
An approach to ==detect specific DNA regions/sequences associated with a protein of interest, in vivo.==
Became a powerful tool to analyze protein/DNA interactions, furthermore to detect any signal/modification associated with DNA/Chromatin.
• Protein-DNA interactions
• Chromatin States
• Transcriptional regulation
• DNA modifications (MeDIP)
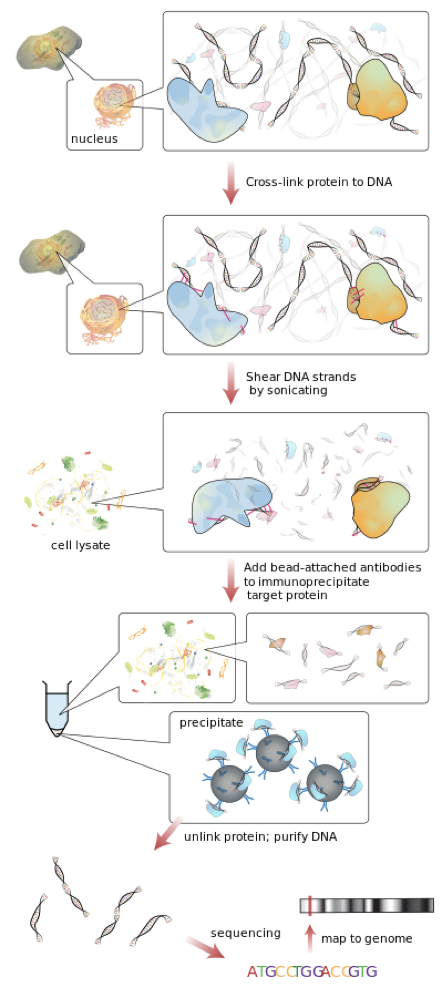
ChIP
•DNA and associated proteins on chromatin in living cells or tissues are crosslinked (this step is omitted in Native ChIP).
•The DNA-protein complexes (chromatin-protein) are then sheared into ~500 bp DNA fragments by sonication or nuclease digestion.
•Cross-linked DNA fragments associated with the protein(s) of interest are selectively immunoprecipitated from the cell debris using an appropriate protein-specific antibody.
•The associated DNA fragments are purified and their sequence is determined. Enrichment of specific DNA sequences represents regions on the genome that the protein of interest is associated with in vivo.
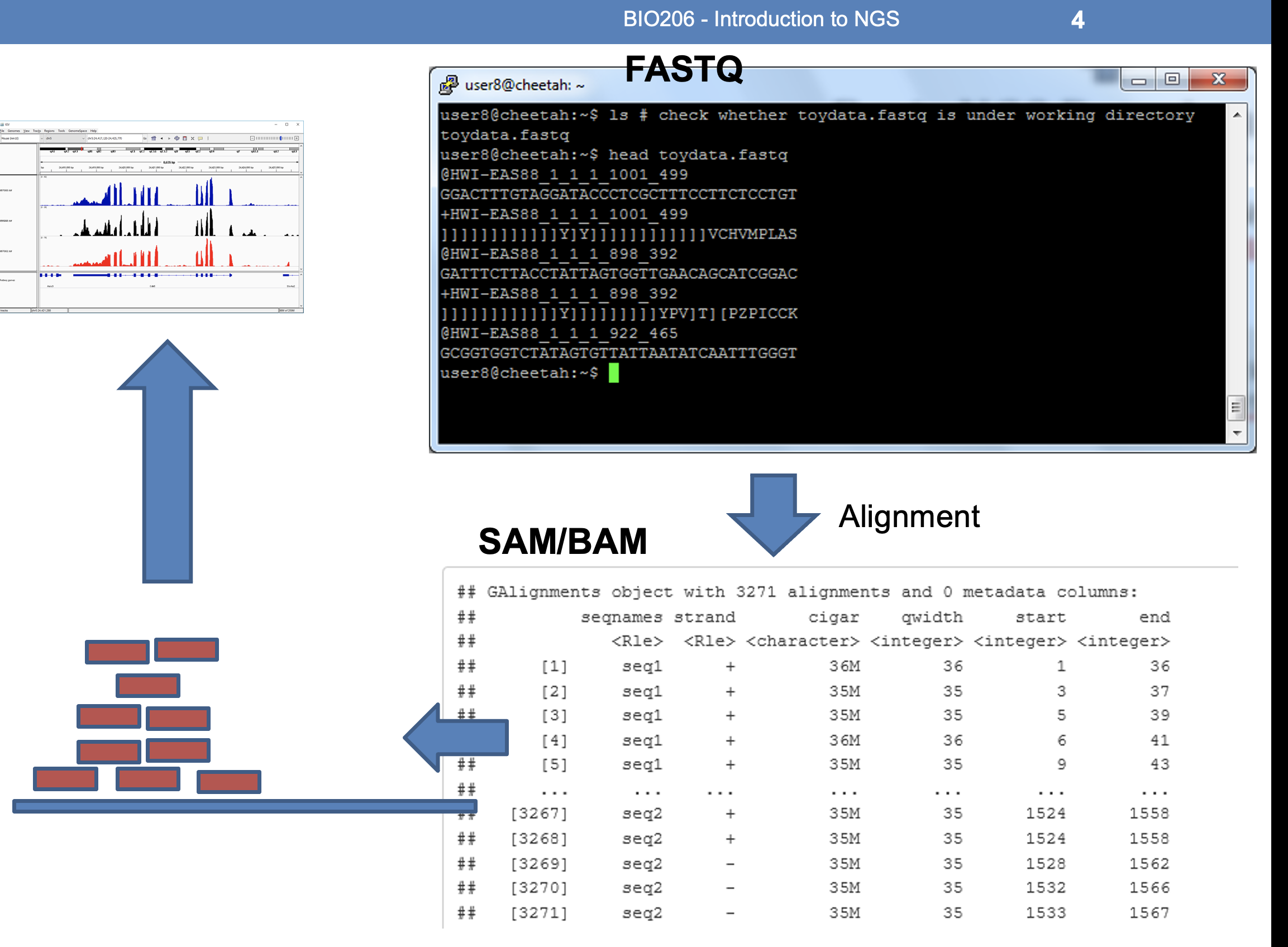
NGS flow chart & details
FASTQ format
SAM format:
SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text format consisting of a header section, which is optional, and an alignment section.
The first line in a FASTA file starts either with a “>” (greater-than) symbol
Following the initial line (used for a unique description of the sequence) is the actual sequence itself in standard one-letter code.
TDF format
A tiled data file (TDF) file (.tdf) is a binary file that contains data that has been preprocessed for faster display in IGV. There are other alternative formats, including wig, bigwig, etc.
TDF: (Aligned) Tag density file for visualization of BAM
GTF: Gene annotation
BED: range format (chr1, 210-350). It can be used to store gene annotation, sequencing read alignment information, TF binding site information, etc.
Key_Info.bash
1 2 3 4 5 6 7 8 9 10 11 12 13
# Project #2 # BIO405 # 2021-03-15 # Bash Script
# S1. make your working directory ################################## # connect to the Linux server pwd# check where you are ls # see what do you have mkdir ~/P2 # make a folder for this project under your home directory ~ # "~" means your home directory on the linux server ls ~ # to check whether you have the P2 folder generated under your home directory
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
# S2. download one fastq data # Alternative 1: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA486780&o=acc_s%3Aa (used example) # Alternative 2: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA403831&o=acc_s%3Aa # Most RNAseq datasets were deposited into GEO: https://www.ncbi.nlm.nih.gov/geo/ # try to find a dataset YOU are interested in. Download process can be done in parallel
# download the fastq file mkdir ~/P2/fastq (nohup fastq-dump --split-3 -v SRR7721730 -O ~/P2/fastq > ~/P2/sra/730.out)& ls -l ~/P2/fastq # check what you have now
# Explanations # (nohup)&: means run the command in background mode # --splite-3: if datas are paired-end, they will be divided into two files # SRRxxxxxx: the sample number in GEO # -O: where to save the downloaded sample # > ~/P2/fastq/730.out: where to save the messages received while downloading the file
ls -l ~/P2/fastq # check what you have now
Download the fastq file
Explanations
(nohup)&: means run the command in background mode
–splite-3: if datas are paired-end, they will be divided into two files
SRRxxxxxx: the sample number in GEO
-O: where to save the downloaded sample
> ~/P2/fastq/730.out: where to save the messages received while downloading the file
1 2 3 4
# S3. Top htop -u "$USER"# check the load of the server and the tasks you are running, if you see too many tasks are running, you should not submit additional jobs. q # to quit from htop command killall -u "$USER"# to terminate all your running jobs
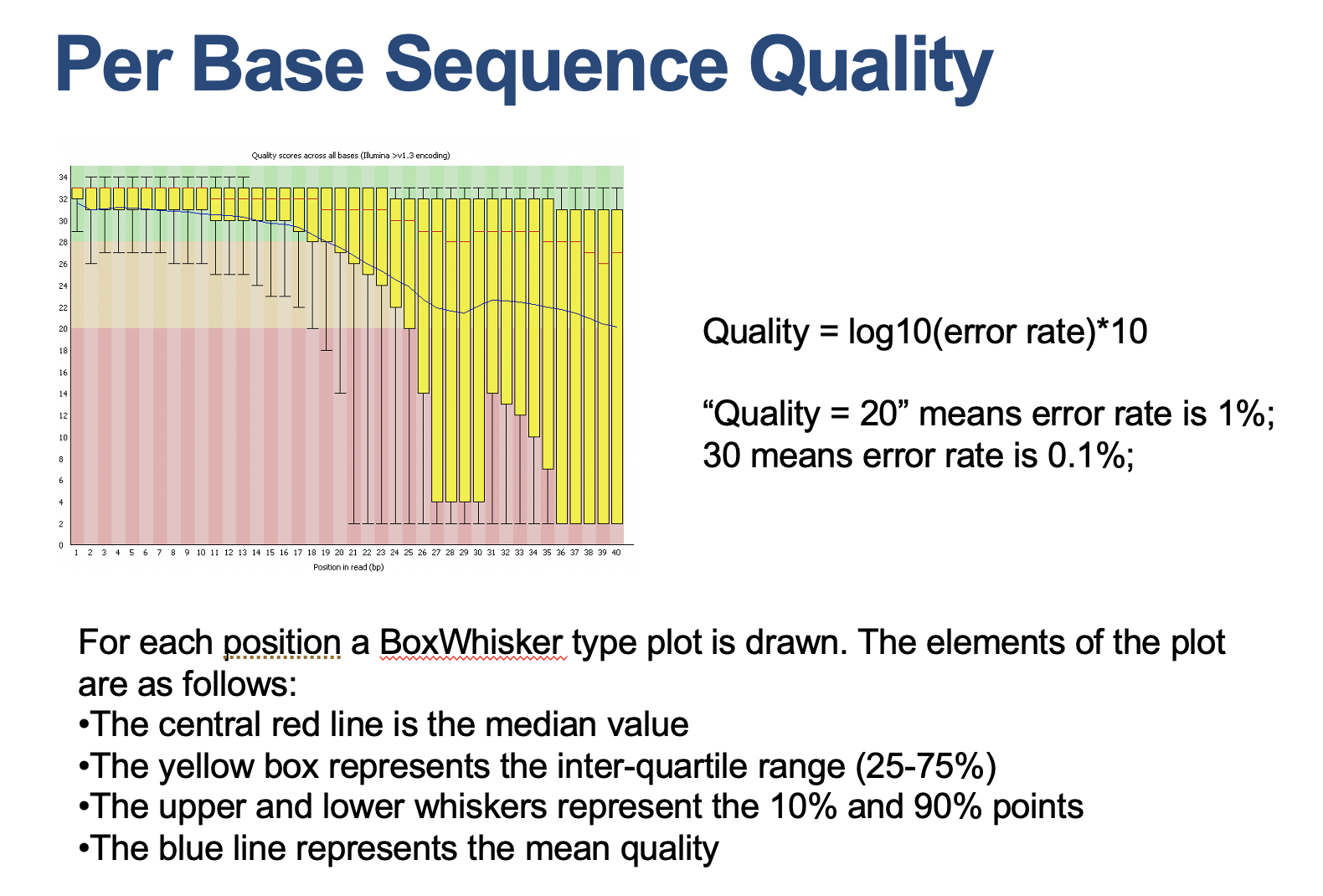
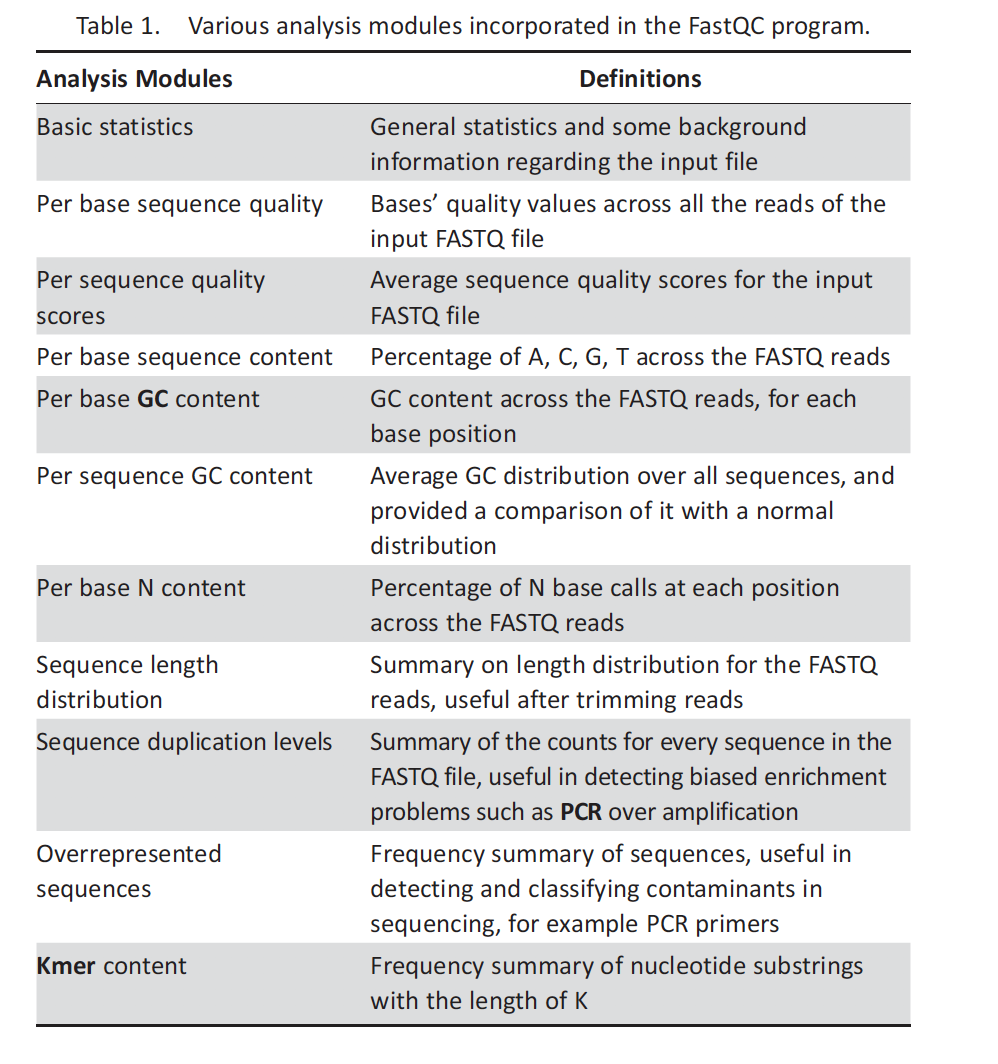
Quality control
1 2 3 4 5 6
# S4. quality control #################################### mkdir ~/P2/fastqc_report fastqc -o ~/P2/fastqc_report -f fastq ~/P2/fastq/SRR7721730_1.fastq ~/P2/fastq/SRR7721730_2.fastq # for paired end of data, you have to check the data quality of both reads
# you could check the html file in the Rstudio
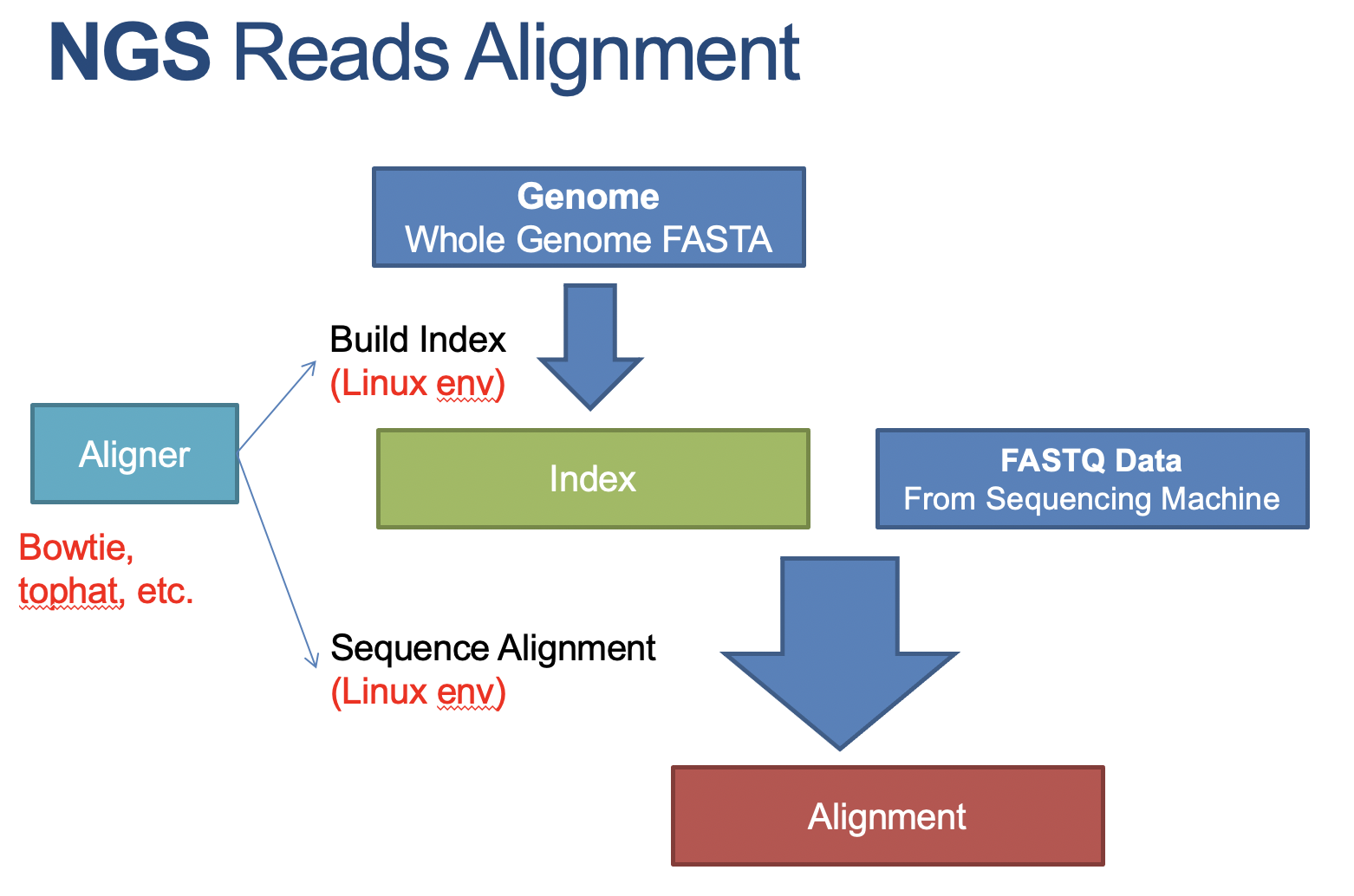
Obtain the index for alignment
1 2 3 4 5 6 7 8 9 10 11 12 13
# S6. get the whole genome fasta mkdir ~/P2/index # it takes a long time to download this genome annotation pacakge, so I have downloaded everything for you. # all the genome annotation (human, mouse, etc) is available from: http://10.7.88.74/MyWeb/Genomes wget -O ~/P2/index/hg38.fa http://10.7.88.74/MyWeb/Genomes/hg38/genome.fa # download genome wget -O ~/P2/index/hg38.gtf http://10.7.88.74/MyWeb/Genomes/hg38/genes.gtf # download annotation
# S7. get the index, needed for alignment # hisat2-build hg38.fa hg38 # build index, take a very long time # with the assistance of index, we could increase the speed of query data (nohup hisat2-build ~/P2/index/hg38.fa ~/P2/index/hg38 > ~/P2/index/index_message.out)& # let us do it in background mode ls -l ~/P2/index/ # the index files are very larger
tophat alignment
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
# S8. Align the fastq files to genome with tophat cd .. mkdir ~/P2/bam # make a folder to store aligned bam files
(nohup hisat2 -x ~/P2/index/hg38 -1 ~/P2/fastq/SRR7721730_1.fastq -2 ~/P2/fastq/SRR7721730_2.fastq -S ~/P2/bam/SRR7721730.sam > ~/P2/bam/hisat_730.out)& head ~/P2/bam/SRR7721730/730.out # check the summary result of alignment
# covert to bam file to save space (nohup samtools view -S ~/P3/bam/SRR7721730.sam -b > ~/P3/bam/SRR7721730.bam)&
# index all the bam files, which is required for visualization in IGV (nohup samtools index ~/P3/bam/SRR7721730_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file.
TDF file for visualization of bam files
IGV browser, downloading it when required
1 2 3 4 5 6 7 8 9 10 11
# S9. prepare TDF file for visualization of bam files # download the chromesome size file wget -O ~/P2/tdf/hg38.chrom.sizes http://10.7.88.74/MyWeb/Genomes/chrom.sizes/hg38.chrom.sizes # use a different genome size file if necessary mkdir ~/P2/tdf (nohup igvtools count -z 5 -w 10 -e 0 ~/P2/bam/SRR7721730_sorted.bam ~/P2/tdf/SRR7721730.tdf ~/P2/tdf/hg38.chrom.sizes > ~/P2/tdf/igv_730.out)& ls ~/P2/tdf
# S10. genome browser # download IGV browser: http://software.broadinstitute.org/software/igv/download # use IGV to visualize the TDF files # use IGV to visualize the BAM files (with the .bai index file)
use IGV to visualize the TDF files
use IGV to visualize the BAM files (with the .bai index file)
# S1. make your working directory ################################## # connect to the Linux server cd ~ # go to your home directory mkdir P3 # or use whatever name you prefer ls # check whether you have the new folder for project 3 pwd# check your current working directory
# S2. download the raw data in sra format #################################### # Data from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53794 mkdir ~/P3/fastq # make a new directory to store raw sequencing data in SRA format ls ~/P3/fastq # nothing is inside
# the structure of this command is: (nohup command_to_be_executed > text_log_file.out)& ## --splite-3 for mate-pairs: biological reads satisfying dumping conditions are placed in files *_1.fastq and *_2.fastq. ## If only one biological read is present it is placed in *.fastq Biological reads and above are ignored. ## If the data is single-end, it will not save as two files.
Syntax explanation
the structure of this command is: ==(nohup command_to_be_executed > text_log_file.out)&==
==–splite-3 for mate-pairs: biological reads satisfying dumping conditions are placed in files *_1.fastq and *_2.fastq.==
If only one biological read is present it is placed in *.fastq Biological reads and above are ignored.
If the data is single-end, it will not save as two files.
top # check the download process ps -u j # check you on-going processes, replace "j" with your own user ID "y18u1" ls ~/P3/fastq # check downloaded fastq files cat ~/P3/fastq/226.out # check whether there is any error during the data downloading
# S4. quality control #################################### mkdir ~/P3/report fastqc -o ~/P3/report -f fastq ~/P3/fastq/SRR1068226.fastq ~/P3/fastq/SRR1068227.fastq\ ~/P3/fastq/SRR1068228.fastq ~/P3/fastq/SRR1068229.fastq ~/P3/fastq/SRR1068230.fastq ~/P3/fastq/SRR1068231.fastq # notice, "\" indicates the line is not over yet, continue in the next line. It is equivalent to the following: # this is different from R, in which you can change the line after any meaningful unit. ls ~/P3/report # check the generated fastqc reports, see whether everything is OK. # you may have to trim the data if necessary
# S5. copy the mouse genome information mkdir ~/P3/index # it takes a long time to download this genome annotation pacakge, so I have downloaded everything for you. # all the genome annotation (human, mouse, etc) is available from: http://10.7.88.74/MyWeb/Genomes wget -O ~/P3/index/mm9.fa http://10.7.88.74/MyWeb/Genomes/mm9/genome.fa wget -O ~/P3/index/mm9.gtf http://10.7.88.74/MyWeb/Genomes/mm9/genes.gtf
# S7. Build index, needed for alignment (nohup hisat2-build ~/P3/index/mm9.fa ~/P3/index/mm9 > ~/P3/index/build_index.out)& # build index, take a very long time top # check the progress cat ~/P3/index/build_index.out # check the notifications ls -l ~/P3/index/ # check the index. the index files are very larger
# S8. Align the fastq files to genome with tophat mkdir ~/P3/bam # make a folder to store aligned bam files (nohup hisat2 -x ~/P3/index/mm9 -U ~/P3/fastq/SRR1068226.fastq -S ~/P3/bam/SRR1068226.sam > ~/P3/bam/226.out)& (nohup hisat2 -x ~/P3/index/mm9 -U ~/P3/fastq/SRR1068227.fastq -S ~/P3/bam/SRR1068227.sam > ~/P3/bam/227.out)& (nohup hisat2 -x ~/P3/index/mm9 -U ~/P3/fastq/SRR1068228.fastq -S ~/P3/bam/SRR1068228.sam > ~/P3/bam/228.out)& (nohup hisat2 -x ~/P3/index/mm9 -U ~/P3/fastq/SRR1068229.fastq -S ~/P3/bam/SRR1068229.sam > ~/P3/bam/229.out)& (nohup hisat2 -x ~/P3/index/mm9 -U ~/P3/fastq/SRR1068230.fastq -S ~/P3/bam/SRR1068230.sam > ~/P3/bam/230.out)& (nohup hisat2 -x ~/P3/index/mm9 -U ~/P3/fastq/SRR1068231.fastq -S ~/P3/bam/SRR1068231.sam > ~/P3/bam/231.out)& # for paired-end data, you can use below command (nohup hisat2 -x ~/P2/index/hg38 -1 ~/P2/fastq/SRR7721730_1.fastq -2 ~/P2/fastq/SRR7721730_2.fastq -S ~/P3/bam/SRR7721730.sam> ~/P2/bam/hisat_730.out)& top # check the progress of all tasks cat ~/P3/bam/226.out # check the alignment efficiency
# conver to bam (nohup samtools view -S ~/P3/bam/SRR1068226.sam -b > ~/P3/bam/SRR1068226.bam)& # covert to bam file to save space (nohup samtools view -S ~/P3/bam/SRR1068227.sam -b > ~/P3/bam/SRR1068227.bam)& # covert to bam file to save space (nohup samtools view -S ~/P3/bam/SRR1068228.sam -b > ~/P3/bam/SRR1068228.bam)& # covert to bam file to save space (nohup samtools view -S ~/P3/bam/SRR1068229.sam -b > ~/P3/bam/SRR1068229.bam)& # covert to bam file to save space (nohup samtools view -S ~/P3/bam/SRR1068230.sam -b > ~/P3/bam/SRR1068230.bam)& # covert to bam file to save space (nohup samtools view -S ~/P3/bam/SRR1068231.sam -b > ~/P3/bam/SRR1068231.bam)& # covert to bam file to save space
# index alignment (nohup samtools index ~/P3/bam/SRR1068226_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/P3/bam/SRR1068227_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/P3/bam/SRR1068228_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/P3/bam/SRR1068229_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/P3/bam/SRR1068230_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file. (nohup samtools index ~/P3/bam/SRR1068231_sorted.bam)& # a file with the same name and ended with ".bai" is generated. This is the index of a BAM file.
# S9. find transcribed regions of a single sample mkdir ~/P3/gtf (nohup cufflinks ~/P3/bam/SRR1068226_sorted.bam -o ~/P3/gtf/SRR1068226 > ~/P3/gtf/cufflinks.out)& # reference-based transcriptome reconstruction head ~/P3/gtf/SRR1068226/transcripts.gtf cufflinks --help# check how to do reference-based transcriptome assembly
# S10. visualization of bam files # we use mm9 here, you may check the folder /home/jmeng/App/Genomes/chrom.sizes for other genome assembly numbers, such as, hg19 and mm10. mkdir ~/P3/tdf wget -O ~/P3/tdf/mm9.chrom.sizes http://10.7.88.74/MyWeb/Genomes/chrom.sizes/mm9.chrom.sizes # use a different genome size file if necessary (nohup igvtools count -z 5 -w 10 -e 0 ~/P3/bam/SRR1068226_sorted.bam ~/P3/tdf/SRR1068226.tdf ~/P3/tdf/mm9.chrom.sizes)& (nohup igvtools count -z 5 -w 10 -e 0 ~/P3/bam/SRR1068226_sorted.bam ~/P3/tdf/SRR1068227.tdf ~/P3/tdf/mm9.chrom.sizes)& (nohup igvtools count -z 5 -w 10 -e 0 ~/P3/bam/SRR1068226_sorted.bam ~/P3/tdf/SRR1068228.tdf ~/P3/tdf/mm9.chrom.sizes)& (nohup igvtools count -z 5 -w 10 -e 0 ~/P3/bam/SRR1068226_sorted.bam ~/P3/tdf/SRR1068229.tdf ~/P3/tdf/mm9.chrom.sizes)& (nohup igvtools count -z 5 -w 10 -e 0 ~/P3/bam/SRR1068226_sorted.bam ~/P3/tdf/SRR1068230.tdf ~/P3/tdf/mm9.chrom.sizes)& (nohup igvtools count -z 5 -w 10 -e 0 ~/P3/bam/SRR1068226_sorted.bam ~/P3/tdf/SRR1068231.tdf ~/P3/tdf/mm9.chrom.sizes)& ls tdf
# use IGV to visualize the TDF files # compare /P3/tdf/SRR1068226.tdf with the gtf # file generated ~/P3/gtf/SRR1068226/transcripts.gtf and with reference
# S11. differential analysis with cuffdiff # copy the gene annotation file mm9.gtf to the same directory, or you may provide a full path # gene annotation GTF of mm9 and hg38 are available at directory: /home/j/BIO405/Genomes, choose the one you need: mkdir ~/P3/DGE (nohup cuffdiff -o ~/P3/DGE -p 2 -L WT,CI994\ -b ~/P3/index/mm9.fa -u ~/P3/index/mm9.gtf\ ~/P3/bam/SRR1068226_sorted.bam,~/P3/bam/SRR1068227_sorted.bam,~/P3/bam/SRR1068228_sorted.bam\ ~/P3/bam/SRR1068229_sorted.bam,~/P3/bam/SRR1068230_sorted.bam,~/P3/bam/SRR1068231_sorted.bam\ > ~/P3/DGE/cuffdiff.out)& # the above step will take a long time, so let us do it in background mode
ps -u $USER ls ~/P3/DGE head ~/P3/DGE/cuffdiff.out head ~/P3/DGE/gene_exp.diff
# Use "Cuffdiff" to conduct differential expression analysis # please check the meaning of "-p 4", "-L WT,CI994", "-b ~/P3/index/mm9.fa", "-u" with the following command cuffdiff --help # check the files generated in output folder DGE specified by "-o ~/P3/DGE"
# S1. MACS2 peak calling from ChIP-seq data # Download ChIPseq data mkdir ~/P4 mkdir ~/P4/fastq mkdir ~/P4/index mkdir ~/P4/report mkdir ~/P4/bam mkdir ~/P4/tdf mkdir ~/P4/macs
# download the data from GEO # please use the data of your interests in the assignment # it is better to find a data set related to your master thesis (nohup fastq-dump SRR568477 -O ~/P4/fastq > ~/P4/fastq/477.out)& (nohup fastq-dump SRR1768320 -O ~/P4/fastq > ~/P4/fastq/320.out)&
# please note that # if the sample use pair-end sequencing, if you should use "fastq-dump --split-files ..." and will get two fastq files. # if the reads quality of your data is weird, it might because you have two reads cancatenated together. # more reads are no pair-end, it means you will see "--split-files" more often in the future.
# check the data quality (nohup fastqc -o ~/P4/report -f fastq ~/P4/fastq/SRR568477.fastq > ~/P4/report/fastqc_477.out)& # quality control (nohup fastqc -o ~/P4/report -f fastq ~/P4/fastq/SRR1768320.fastq > ~/P4/report/fastqc_320.out)& # quality control
# download the genome and annotation file # it takes a long time to do that, so I have downloaded everything for you. # all the genome annotation (human, mouse, etc) is available from: http://10.7.88.74/MyWeb/Genomes wget -O ~/P4/index/mm10.fa http://10.7.88.74/MyWeb/Genomes/mm10/genome.fa # download the genome wget -O ~/P4/index/mm10.gtf http://10.7.88.74/MyWeb/Genomes/mm10/genes.gtf # download the annotation # If you need to work on other species, then you will have to find them from other places.
# you should usually go through the following # build index, needed for alignment (nohup hisat2-build ~/P4/index/mm10.fa ~/P4/index/mm10 > ~/P3/index/build_index.out)& # build index, take a very long time top # check the progress ps -u $USER# check your on-going processes cat ~/P4/index/build_index.out # check the notifications ls -l ~/P4/index/ # check the index. the index files are very larger
# alignment (nohup hisat2 -x ~/P4/index/mm10 -U ~/P4/fastq/SRR568477.fastq -S ~/P4/bam/SRR568477.sam > ~/P4/bam/align_SRR568477.log)& # do alignment in background (nohup hisat2 -x ~/P4/index/mm10 -U ~/P4/fastq/SRR1768320.fastq -S ~/P4/bam/SRR1768320.sam > ~/P4/bam/align_SRR1768320.log)& # do alignment in background
# convert SAM to BAM (nohup samtools view -bS ~/P4/bam/SRR568477.sam -o ~/P4/bam/SRR568477.bam)& # convert sam to bam file (nohup samtools view -bS ~/P4/bam/SRR1768320.sam -o ~/P4/bam/SRR1768320.bam)& # convert sam to bam file ls -l ~/P4/bam/ # check the size of files. BAM is a lot smaller than SAM
# sort the alignments, which is needed for many tools (nohup samtools sort ~/P4/bam/SRR568477.bam -o ~/P4/bam/SRR568477_sorted.bam > ~/P4/bam/sort_SRR568477.out)& # sort alignment by coordinates (nohup samtools sort ~/P4/bam/SRR1768320.bam -o ~/P4/bam/SRR1768320_sorted.bam > ~/P4/bam/sort_SRR1768320.out)& # sort alignment by coordinates
# peak calling (nohup macs2 callpeak -t ~/P4/bam/SRR568477_sorted.bam -c ~/P4/bam/SRR1768320_sorted.bam --outdir ~/P4/macs -n H3K4me3 -g mm -f BAM -q 0.01 > ~/P4/macs/callpeak.out)& # peak calling # load the .BED file in IGV # compare the peaks called with tdf file and gene annotation information in IGV
# motif finding (nohup bedtools getfasta -fi ~/P4/index/mm10.fa -bed ~/P4/macs/H3K4me3_peaks.narrowPeak -fo ~/P4/macs/H3K4me3_region.fa > ~/P4/macs/findmotif.out)& # extract sequence # extract the sequence for motif finding: https://meme-suite.org/meme/tools/streme # if the file H3K4me3_region.fa is too large, you may use the first 10000 lines # head -n 10000 ~/P4/macs/H3K4me3_region.fa > ~/P4/macs/H3K4me3_region_top_10000.fa
### A little advanced bash programming. # This part is not related to the ChIPseq data analysis practical session # Use "Ctrl + c" to terminate a running job # You can run tasks in the background. If the tasks are running the background, you can log out and leave with the tasks still running on the server.
Sequence data analysis
- Determination of binding sites from the sequence data is a challenge. Conceptually, genomic regions with an increased number of sequencing reads (tags) compared to control is considered to be a TFBS
- Statistical filtering criteria is used to determine if these putative sites represent true binding sites.
- After statistical analysis of binding sites a further analysis of data is required. These may include analysis of location of binding, relative to transcription factor binding sites or potential nearby target genes.
cuffdiff v2.2.1 () ----------------------------- Usage: cuffdiff [options] <transcripts.gtf> <sample1_hits.sam> <sample2_hits.sam> [... sampleN_hits.sam] Supply replicate SAMs as comma separated lists for each condition: sample1_rep1.sam,sample1_rep2.sam,...sample1_repM.sam General Options: -o/--output-dir write all output files to this directory [ default: ./ ] -L/--labels comma-separated list of condition labels --FDR False discovery rate used in testing [ default: 0.05 ] -M/--mask-file ignore all alignment within transcripts in this file [ default: NULL ] -C/--contrast-file Perform the constrasts specified in this file [ default: NULL ] -b/--frag-bias-correct use bias correction - reference fasta required [ default: NULL ] -u/--multi-read-correct use 'rescue method'for multi-reads [ default: FALSE ] -p/--num-threads number of threads used during quantification [ default: 1 ] --no-diff Don't generate differential analysis files [ default: FALSE ] --no-js-tests Don't perform isoform switching tests [ default: FALSE ] -T/--time-series treat samples as a time-series [ default: FALSE ] --library-type Library prep used for input reads [ default: below ] --dispersion-method Method used to estimate dispersion models [ default: below ] --library-norm-method Method used to normalize library sizes [ default: below ]
Advanced Options: -m/--frag-len-mean average fragment length (unpaired reads only) [ default: 200 ] -s/--frag-len-std-dev fragment length std deviation (unpaired reads only) [ default: 80 ] -c/--min-alignment-count minimum number of alignments in a locus for testing [ default: 10 ] --max-mle-iterations maximum iterations allowed for MLE calculation [ default: 5000 ] --compatible-hits-norm count hits compatible with reference RNAs only [ default: TRUE ] --total-hits-norm count all hits for normalization [ default: FALSE ] -v/--verbose log-friendly verbose processing (no progress bar) [ default: FALSE ] -q/--quiet log-friendly quiet processing (no progress bar) [ default: FALSE ] --seed value of random number generator seed [ default: 0 ] --no-update-check do not contact server to check for update availability[ default: FALSE ] --emit-count-tables print count tables used to fit overdispersion [ DEPRECATED ] --max-bundle-frags maximum fragments allowed in a bundle before skipping [ default: 500000 ] --num-frag-count-draws Number of fragment generation samples [ default: 100 ] --num-frag-assign-draws Number of fragment assignment samples per generation [ default: 50 ] --max-frag-multihits Maximum number of alignments allowed per fragment [ default: unlim ] --min-outlier-p Min replicate p value to admit for testing [ DEPRECATED ] --min-reps-for-js-test Replicates needed for relative isoform shift testing [ default: 3 ] --no-effective-length-correction No effective length correction [ default: FALSE ] --no-length-correction No length correction [ default: FALSE ] -N/--upper-quartile-norm Deprecated, use --library-norm-method [ DEPRECATED ] --geometric-norm Deprecated, use --library-norm-method [ DEPRECATED ] --raw-mapped-norm Deprecated, use --library-norm-method [ DEPRECATED ] --poisson-dispersion Deprecated, use --dispersion-method [ DEPRECATED ]
Debugging use only: --read-skip-fraction Skip a random subset of reads this size [ default: 0.0 ] --no-read-pairs Break all read pairs [ default: FALSE ] --trim-read-length Trim reads to be this long (keep 5' end) [ default: none ] --no-scv-correction Disable SCV correction [ default: FALSE ] Supported library types: ff-firststrand ff-secondstrand ff-unstranded fr-firststrand fr-secondstrand fr-unstranded (default) transfrags Supported dispersion methods: blind per-condition poisson pooled (default) Supported library normalization methods: classic-fpkm geometric (default) geometric quartile
cuffdiff 生成的文件
==FPKM: isoform水平上的丰度, Fragments Per Kilobase of exon model per Million mapped fragments;==
cufflinks: unrecognized option '--help' cufflinks v2.2.1 linked against Boost version 106501 ----------------------------- Usage: cufflinks [options] <hits.sam> General Options: -o/--output-dir write all output files to this directory [ default: ./ ] -p/--num-threads number of threads used during analysis [ default: 1 ] --seed value of random number generator seed [ default: 0 ] -G/--GTF quantitate against reference transcript annotations -g/--GTF-guide use reference transcript annotation to guide assembly 提供GFF文件,以此来指导转录子组装(RABT assembly)。此时,输出结果会包含reference transcripts和novel genes and isforms。 -M/--mask-file ignore all alignment within transcripts in this file -b/--frag-bias-correct use bias correction - reference fasta required [ default: NULL ] -u/--multi-read-correct use 'rescue method'for multi-reads (more accurate) [ default: FALSE ] --library-type library prep used for input reads [ default: below ] --library-norm-method Method used to normalize library sizes [ default: below ]
Advanced Abundance Estimation Options: -m/--frag-len-mean average fragment length (unpaired reads only) [ default: 200 ] -s/--frag-len-std-dev fragment length std deviation (unpaired reads only) [ default: 80 ] --max-mle-iterations maximum iterations allowed for MLE calculation [ default: 5000 ] --compatible-hits-norm count hits compatible with reference RNAs only [ default: FALSE ] --total-hits-norm count all hits for normalization [ default: TRUE ] --num-frag-count-draws Number of fragment generation samples [ default: 100 ] --num-frag-assign-draws Number of fragment assignment samples per generation [ default: 50 ] --max-frag-multihits Maximum number of alignments allowed per fragment [ default: unlim ] --no-effective-length-correction No effective length correction [ default: FALSE ] --no-length-correction No length correction [ default: FALSE ] -N/--upper-quartile-norm Deprecated, use --library-norm-method [ DEPRECATED ] --raw-mapped-norm Deprecated, use --library-norm-method [ DEPRECATED ]
Advanced Assembly Options: -L/--label assembled transcripts have this ID prefix [ default: CUFF ] -F/--min-isoform-fraction suppress transcripts below this abundance level [ default: 0.10 ] -j/--pre-mrna-fraction suppress intra-intronic transcripts below this level [ default: 0.15 ] -I/--max-intron-length ignore alignments with gaps longer than this [ default: 300000 ] -a/--junc-alpha alpha for junction binomial test filter [ default: 0.001 ] -A/--small-anchor-fraction percent read overhang taken as 'suspiciously small' [ default: 0.09 ] --min-frags-per-transfrag minimum number of fragments needed for new transfrags [ default: 10 ] --overhang-tolerance number of terminal exon bp to tolerate in introns [ default: 8 ] --max-bundle-length maximum genomic length allowed for a given bundle [ default:3500000 ] --max-bundle-frags maximum fragments allowed in a bundle before skipping [ default: 500000 ] --min-intron-length minimum intron size allowed in genome [ default: 50 ] --trim-3-avgcov-thresh minimum avg coverage required to attempt 3' trimming [ default: 10 ] --trim-3-dropoff-frac fraction of avg coverage below which to trim 3' end [ default: 0.1 ] --max-multiread-fraction maximum fraction of allowed multireads per transcript [ default: 0.75 ] --overlap-radius maximum gap size to fill between transfrags (in bp) [ default: 50 ]
Advanced Reference Annotation Guided Assembly Options: --no-faux-reads disable tiling by faux reads [ default: FALSE ] --3-overhang-tolerance overhang allowed on 3' end when merging with reference[ default: 600 ] --intron-overhang-tolerance overhang allowed inside reference intron when merging [ default: 30 ] Advanced Program Behavior Options: -v/--verbose log-friendly verbose processing (no progress bar) [ default: FALSE ] -q/--quiet log-friendly quiet processing (no progress bar) [ default: FALSE ] --no-update-check do not contact server to check for update availability[ default: FALSE ] Supported library types: ff-firststrand ff-secondstrand ff-unstranded fr-firststrand fr-secondstrand fr-unstranded (default) transfrags Supported library normalization methods: classic-fpkm
当使用cufflinks或cuffdiff出现了“crash with a ‘bad_alloc’ error”,cuffdiff和cufflinks运行了很长时间才结束————这表明计算机拼接一个高表达的基因或定量分析一个高表达的基因,运行的内存使用玩尽了!解决方法:修改选项“-max-bundle-frags”,可以先尝试500000,若错误依旧在,可以继续下调!
You are using Cufflinks v2.2.1, which is the most recent release. open: No such file or directory File 30 doesn’t appear to be a valid BAM file, trying SAM… Error: cannot open alignment file 30 for reading 这表明,你的参数有问题。例如“–min-intron-length”,你设置为了:“-min-intron-length”
Cufflinks输出结果
cufflinks的输入文件是sam或bam格式。并且sam或bam格式的文件必须排好序。(The SAM file supplied to Cufflinks must be sorted by reference position.)