- 研究tutorial2 的Tandem MS-Sequence Confirmation
- 看视频
- 还是需要研究Mass spectrometry
Proteomics workflow
1.Extract proteins from sample
2.Separate proteins (various techniques)
3.Digest proteins into peptides
4.Separate peptides (various techniques)
5.Analyse peptides by mass spectrometry (MS)
e.g. known masses of peptides or masses of peptide fragments
6.Database search to identify peptides (and hence protein)
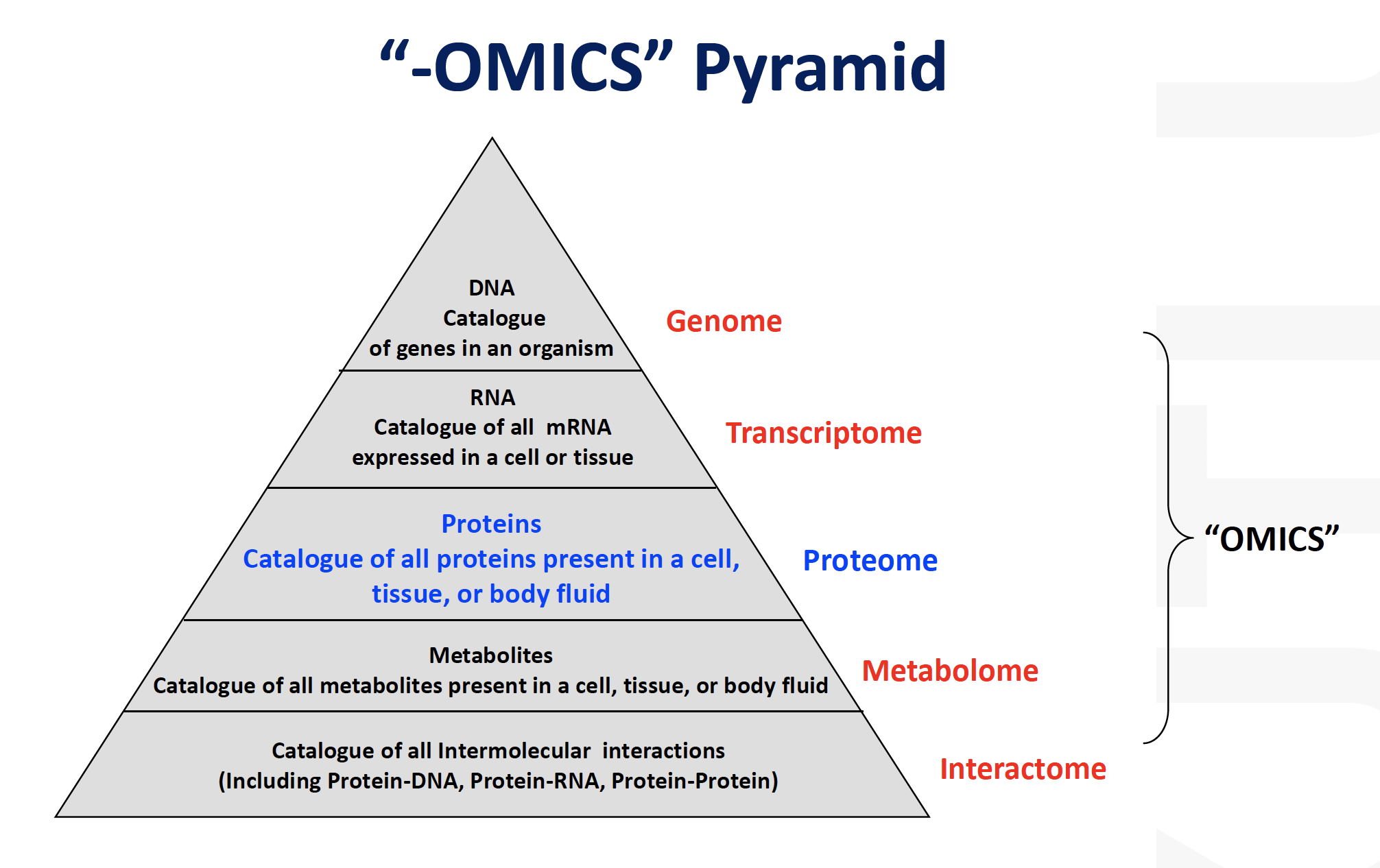
Proteomics
- Complex mixtures
- Partial sequence analysis
- Emphasis in identification by database matching
- Systems biology

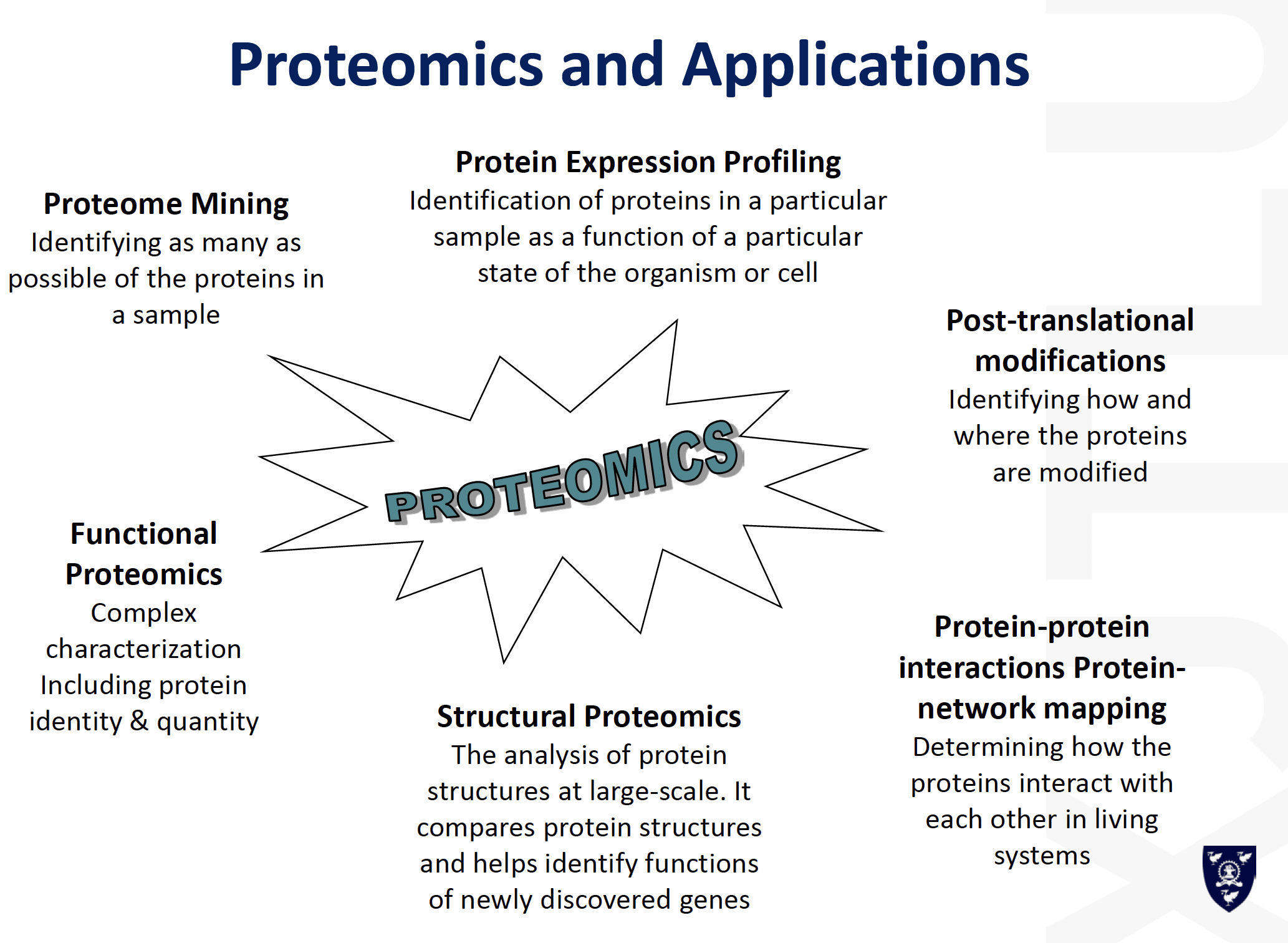
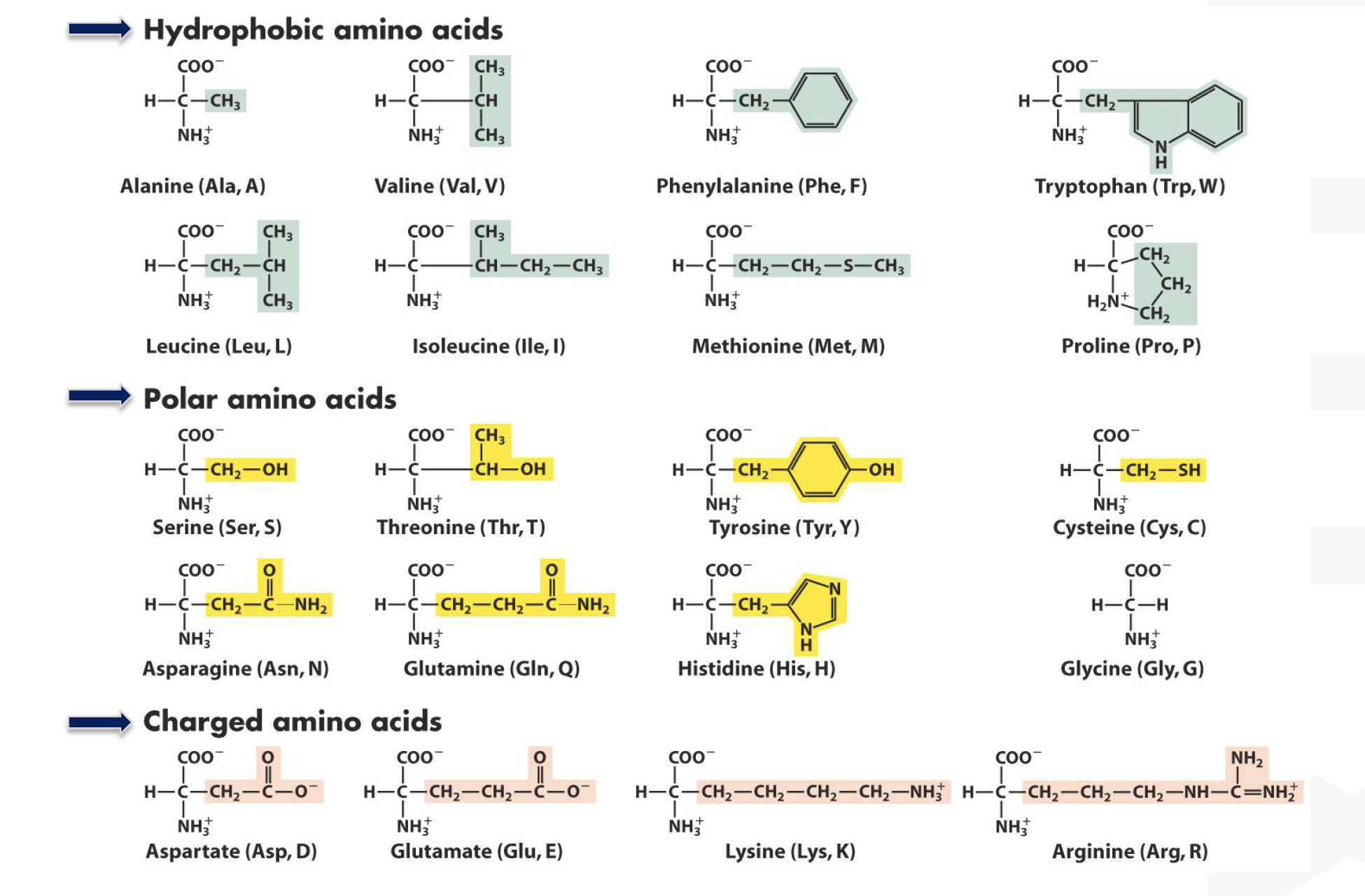
Mass Spectrometry
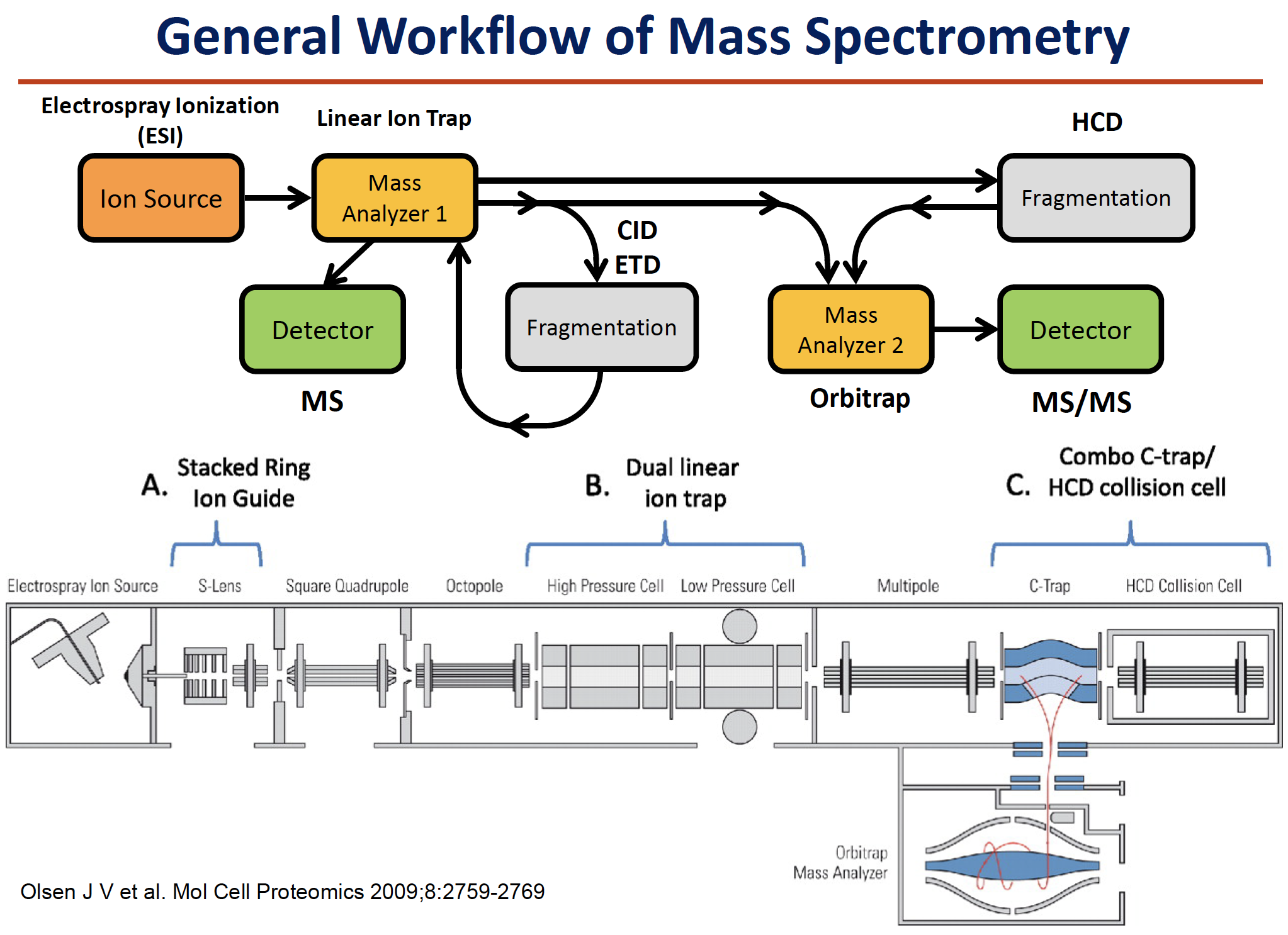
Mass Spectrometry-based Protein Identification
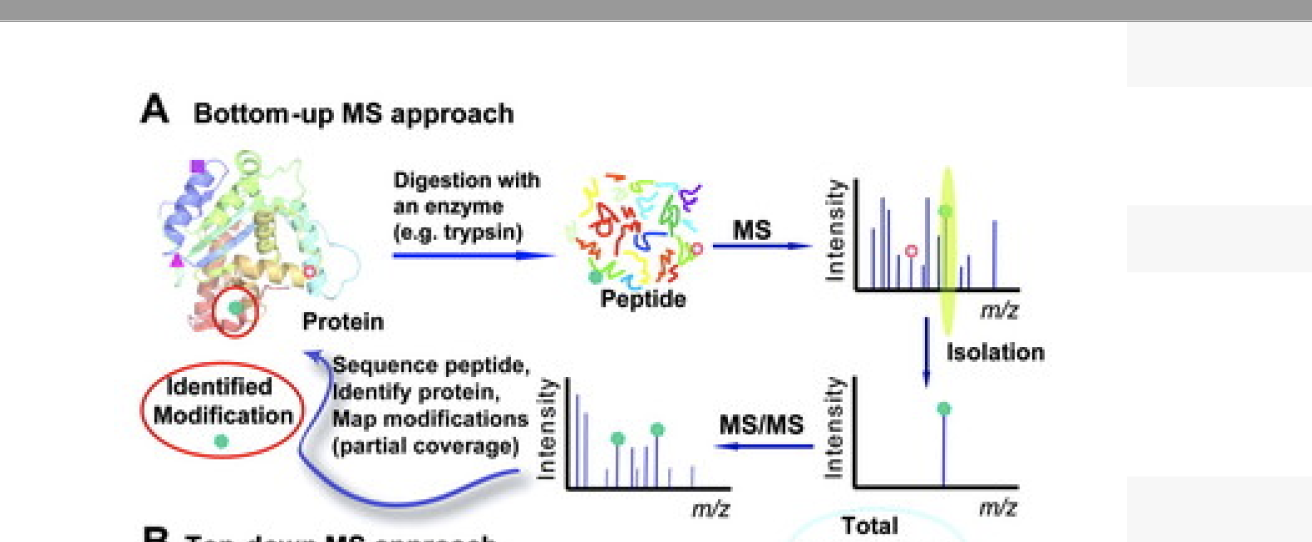
- A mass spectrum of the resulting digest products produces a “peptide map”or a “peptide fingerprint”.
The measured masses can be compared to theoretical peptide maps derived from database sequences for identification. There are a few choices of mass analysis that can be selected from this point, depending on available instrumentation and other factors. The resulting peptide fragments can be subjected to
MALDI-MSor ESI-MSanalysis.
- For example, a small aliquot of the digest solution can be directly analyzed by MALDI-MS to obtain a peptide map. The resulting sequence coverage (relative to the entire protein sequence) displayed from the total number of tryptic peptides observed in the MALDI mass spectrum can be quite high, i.e., greater than 80% of the sequence, although it can vary considerably depending on the protein, sample amount, etc. The measured molecular weights of the peptide fragments along with the specificity of the enzyme employed can be searched and compared against protein sequence databasesusing a number of searching algorithms available either commercially or on the Internet.
cw1
Task1
What is a proteome and how is the link between an organism’s genome and a proteome? (10 marks)
Proteome represents the proteins catalogue of all proteins in a cell tissue or body fluid. Transcriptome, which is the RNA catalogue of all mRNA expressed in a cell or tissue, is the link between an organisms genome and proteome.
Task2
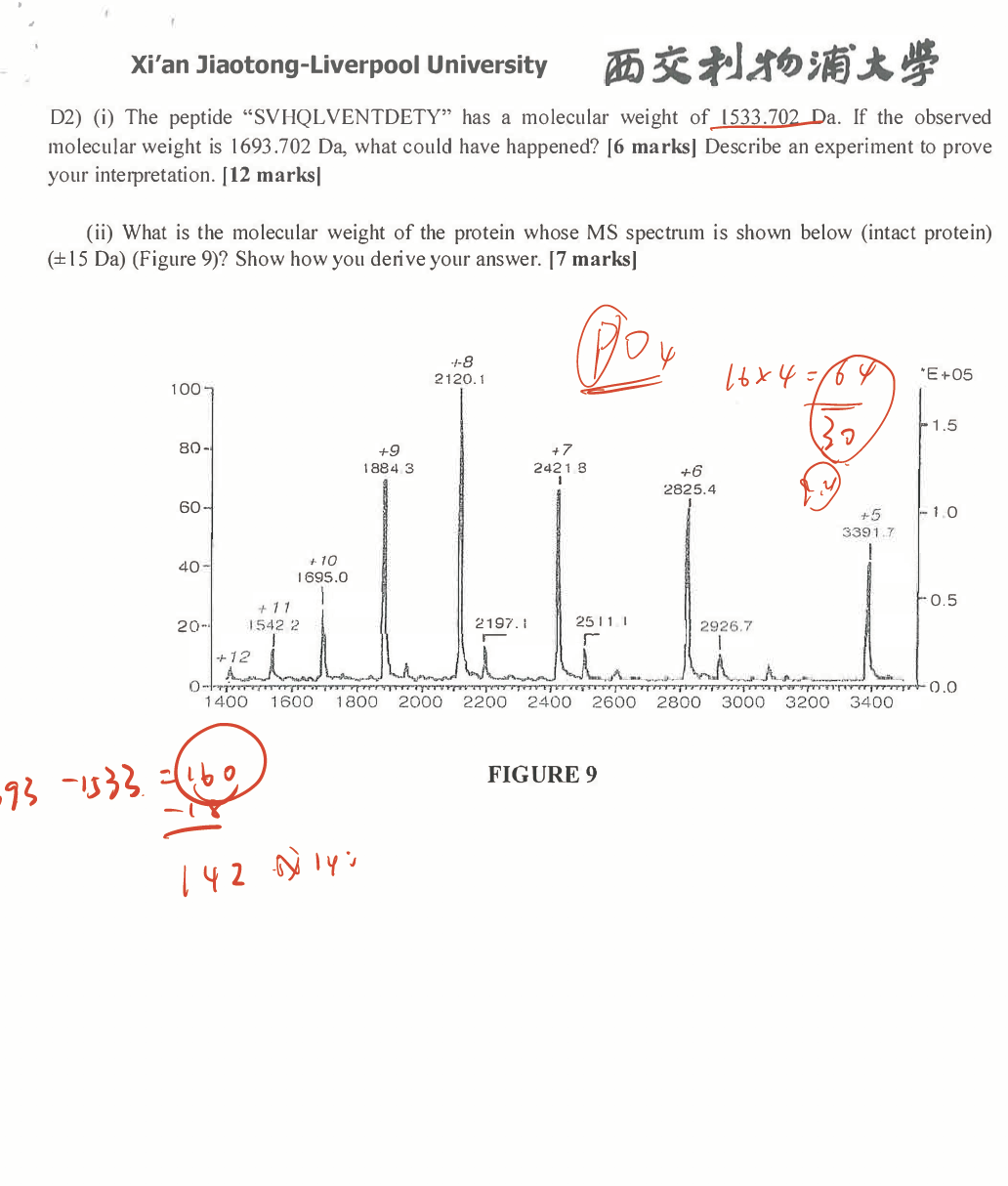
An investigator brought a purified peptide to the Proteomics Core Lab for MALDI-MS analysis for quality control purpose. The peptide was dissolved in a buffer containing 10mM Tris-HCl, pH 7.5, 250mM NaCl, and trace amount of NP40. The theoretical molecular weight of the peptide should be 1571.5Da. However, the MALDI-MS spectrum showed two major peaks, one at m/z=1572.6, and another at m/z=1594.6. Did the investigator have the right peptide? If yes, is the peptide relatively pure? Explain why. (10 marks)
Theoretical: 1571
Results: 1572.6; 1594.6
The investigator could have the right, pure peptide, but require further sequence analysis confirmation. The possible reasons for the situation could from two aspects. One is the difference in digestion efficiency, some peptide may suffer from different lysing rate and therefore the different peak; another one could be the situation that some peptide might be ionized during MALDI process, leading to the different major peaks. Based on the previous analysis, we could not jump to the conclusion, further analysis would be required.
MALDI-MS:
==Matrix-Assisted Laser Desorption/Ionization (MALDI)==
可能的答案:


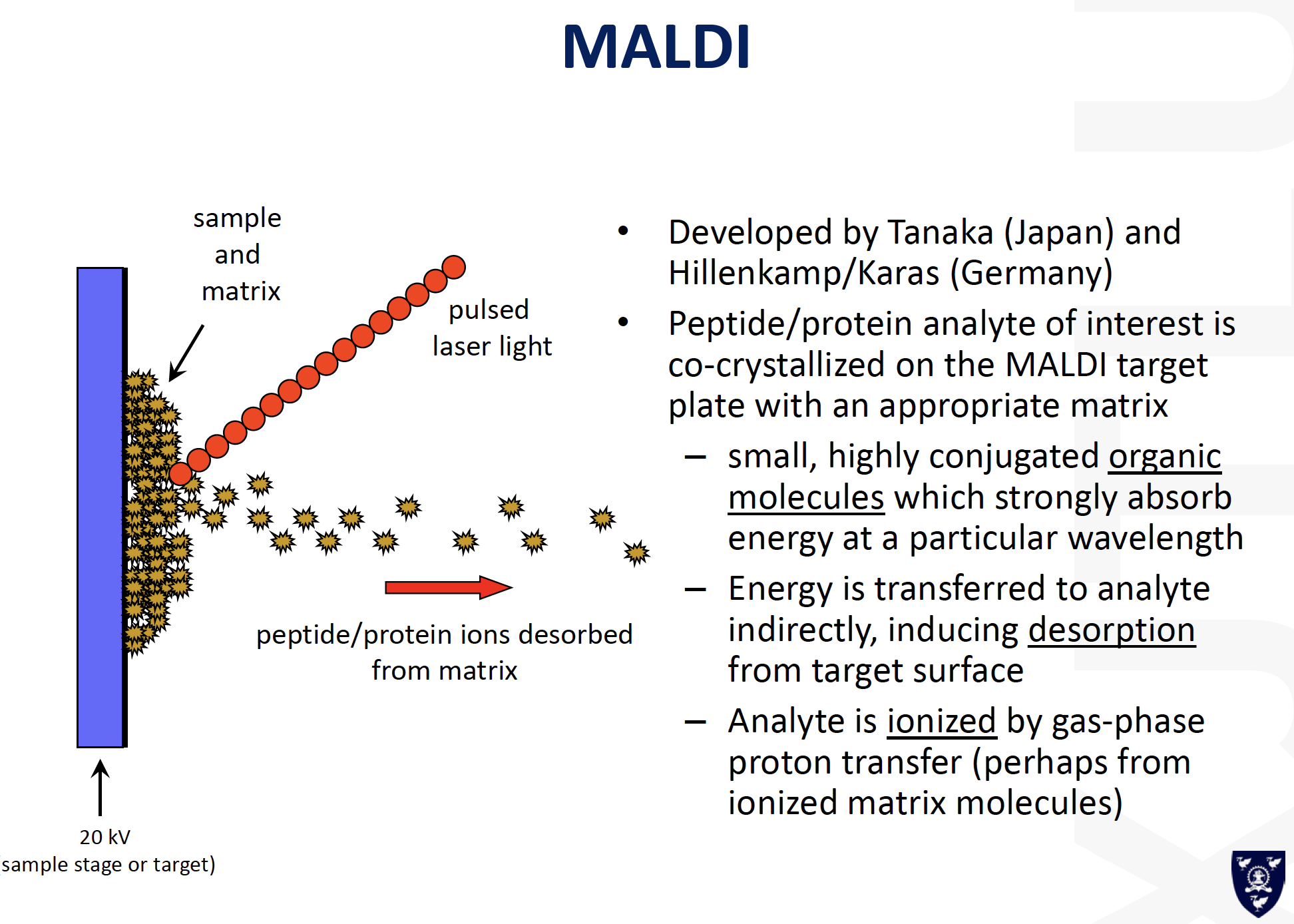
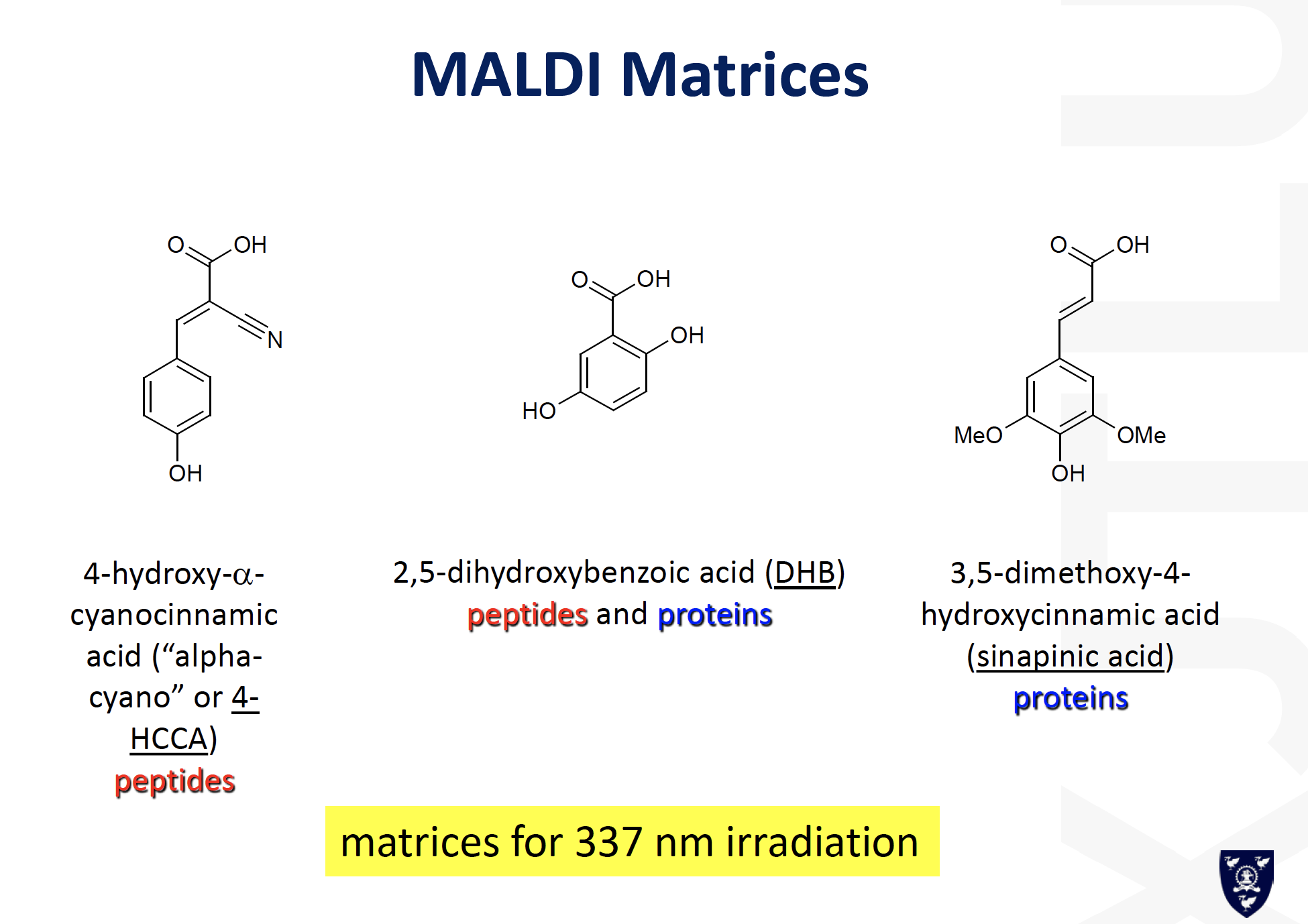
Task3
BRCA1 is a breast cancer related protein. However, in many breast cancer biomarker discovery studies, BRCA1 cannot be identified by general shotgun LC/MS-based proteomics approaches which use RP-LC to separate tryptic peptides. Its abundance in cancer cells should be sufficient enough to be detected by the currently available mass spectrometers such as Orbitrap FusionTM. Please describe the most likely reason for the failure of its detection (please keep your answers within one-page limit). (20 marks)
Why LC/MS-based proteomics approaches which use RP-LC to separate tryptic peptides failed?
==RP-LC:== reversed phase

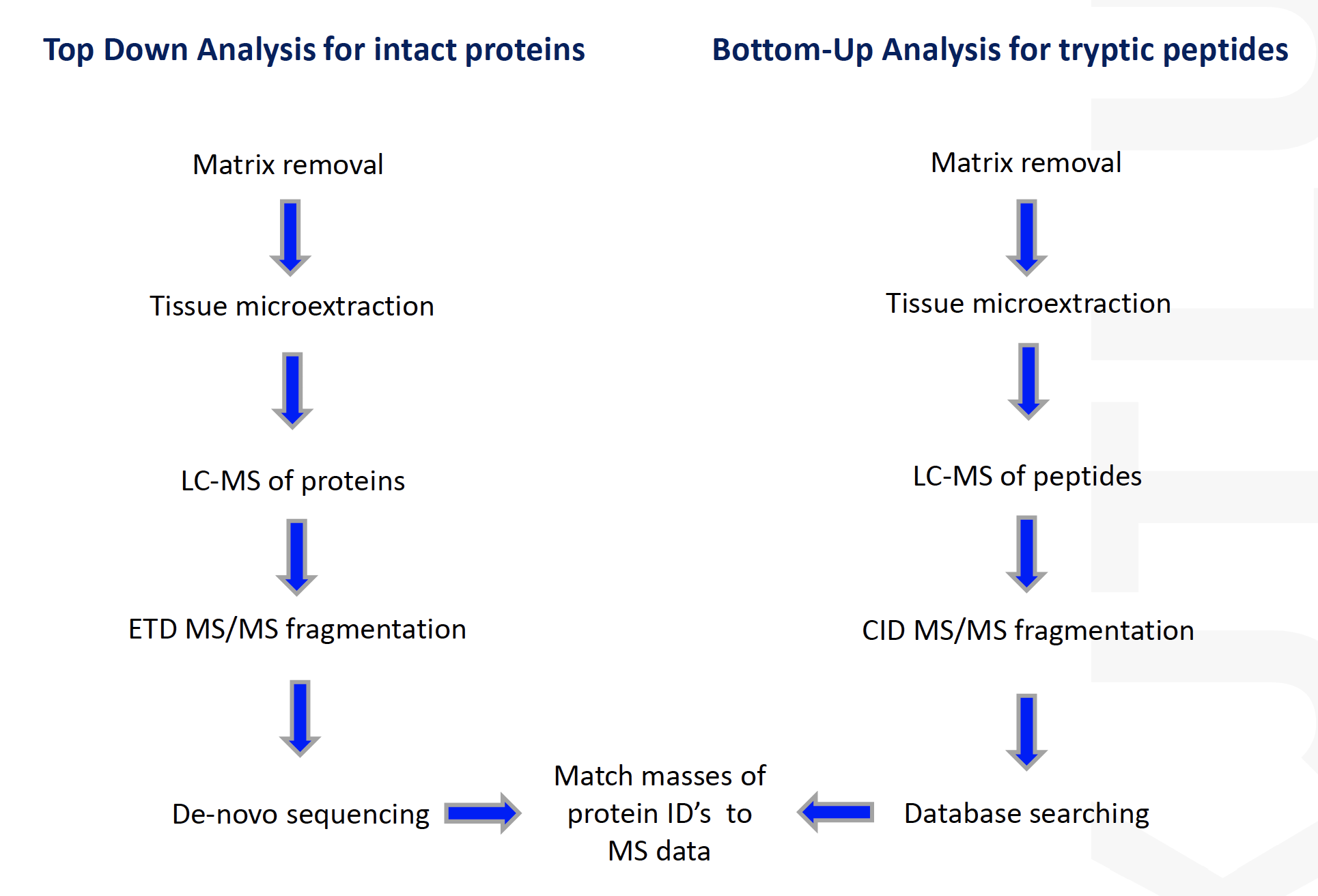
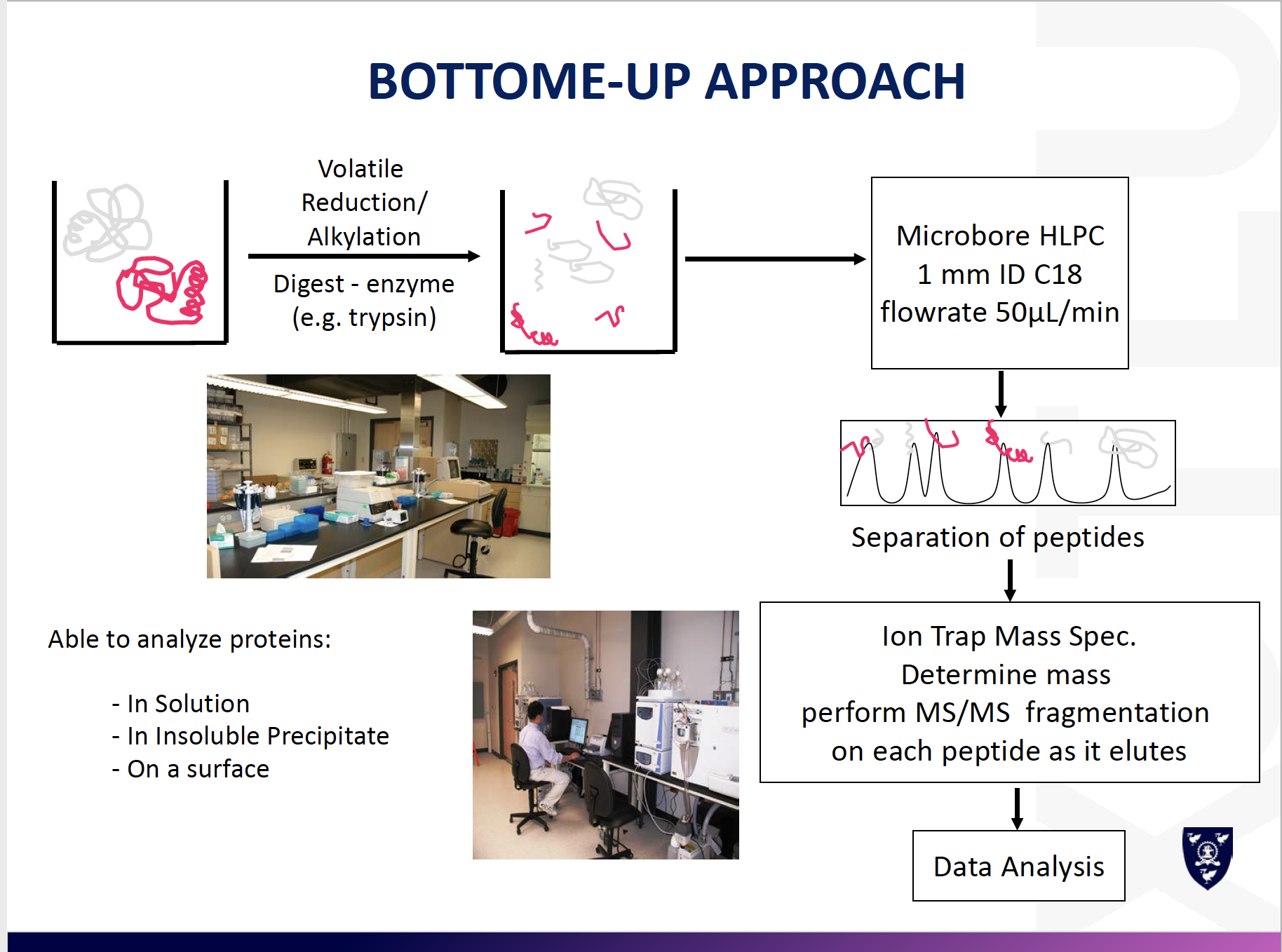
Bottom-Up: sequence tryptic fragments of larger proteins
- Easier to analyze larger or hydrophobic proteins
- Potential for MALDI MS/MS
- Analysis of proteins from their derived peptides in complex biological systems
- LC-MS/MS (generally OrbitrapMS) is the technique of choice
网络上的消息
Phowever, identification of clinical biomarkers remains one of the most challenging applications [33]. Current biomarkers or biomarker candidates struggle with limited reliability and proper validation as well as with limited sensitivity and specificity [32].
There are several important factors to be considered in the search for a biomarker [54]. First of all,
tissues as samples are not easily accessible, and, as they are composed of different cells, they are also very heterogeneous, an issue being addressed with labour intensive laser capture microdissection.
its complexity, time consumption, the need for educating the personnel, the costs associated with purchase of equipment, and last but not least, the availability of tissue samples. In clinical diagnosis there has always been a tendency to use most easily accessible specimens for diagnostic procedures, such as blood, saliva, urine, and faeces.
dynamic complexity and are dependent on patient and environmental characteristics.
collecting the proper number of patients for biomarker validation can also pose a problem.
And finally, it is becoming obvious that a single biomarker is not enough for accurate screening or diagnostic purposes;
rather a panel of proteins will be necessary
Global LC-MS/MS-based proteomics suffers inherent problems of:
quantification accuracy and reproducibility (i.e., missing data in samples with low-abundance proteins that fail to be identified) in large-scale studies due to the stochastic sampling nature of shotgun MS/MS
Reversed-Phase Liquid Chromatography
Reversed-Phase Liquid Chromatography (RPLC) is one of the High Pressure Liquid Chromatography or “HPLC” approaches, which are now for commercial reasons often alluded to as High Performance Liquid Chromatography.
RPLC is especially useful in the separation and purification of proteins. To that effect columns are used which are packed with, e.g., porous glass beads that are made hydrophobic by treating them with octadecyl (C18) groups.
In water such C18-coated beads bind hydrophobically to all but the most hydrophilic proteins.
For the subsequent elution of the bound proteins one decreases the hydrophobizing capability of the aqueous medium by the admixture of water-miscible organic solvents with much lower AB values than the 51 mJ/m2 of water, such as methanol, ethanol, ethylene glycol, or acetonitrile.
正儿八经的answer
RP-LC, the abbreviation of Reversed-Phase Liquid Chromatography, is especially useful in the separation and purification of proteins. The hydrophobic porous beads with octadecyl (C18) groups could bind hydrophobically to all but the most hydrophilic proteins, enabling the separation of proteins.
As mentioned in the questions that BRCA1could not be identified by general shotgun LC/MS-based proteomics approaches which use RP-LC to separate tryptic peptides, but work in the mass spectrometers. Therefore, I could hypothesis that the most likely reason could related to the hydrophobicity of BRCA1 biomarker.
Current biomarkers identification struggled with limited reliability and proper validation as well as with limited sensitivity and specificity. Since biomarkers are highly dependent on environmental characteristics, which could be easily influenced by surrounding environment
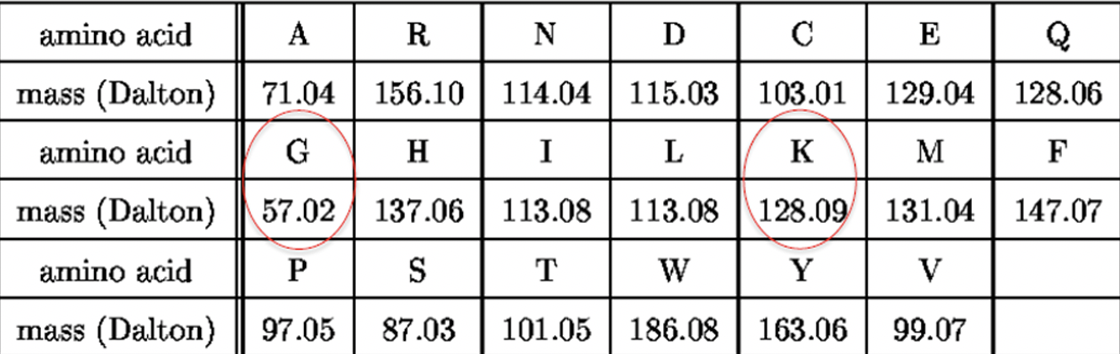
建议使用的table
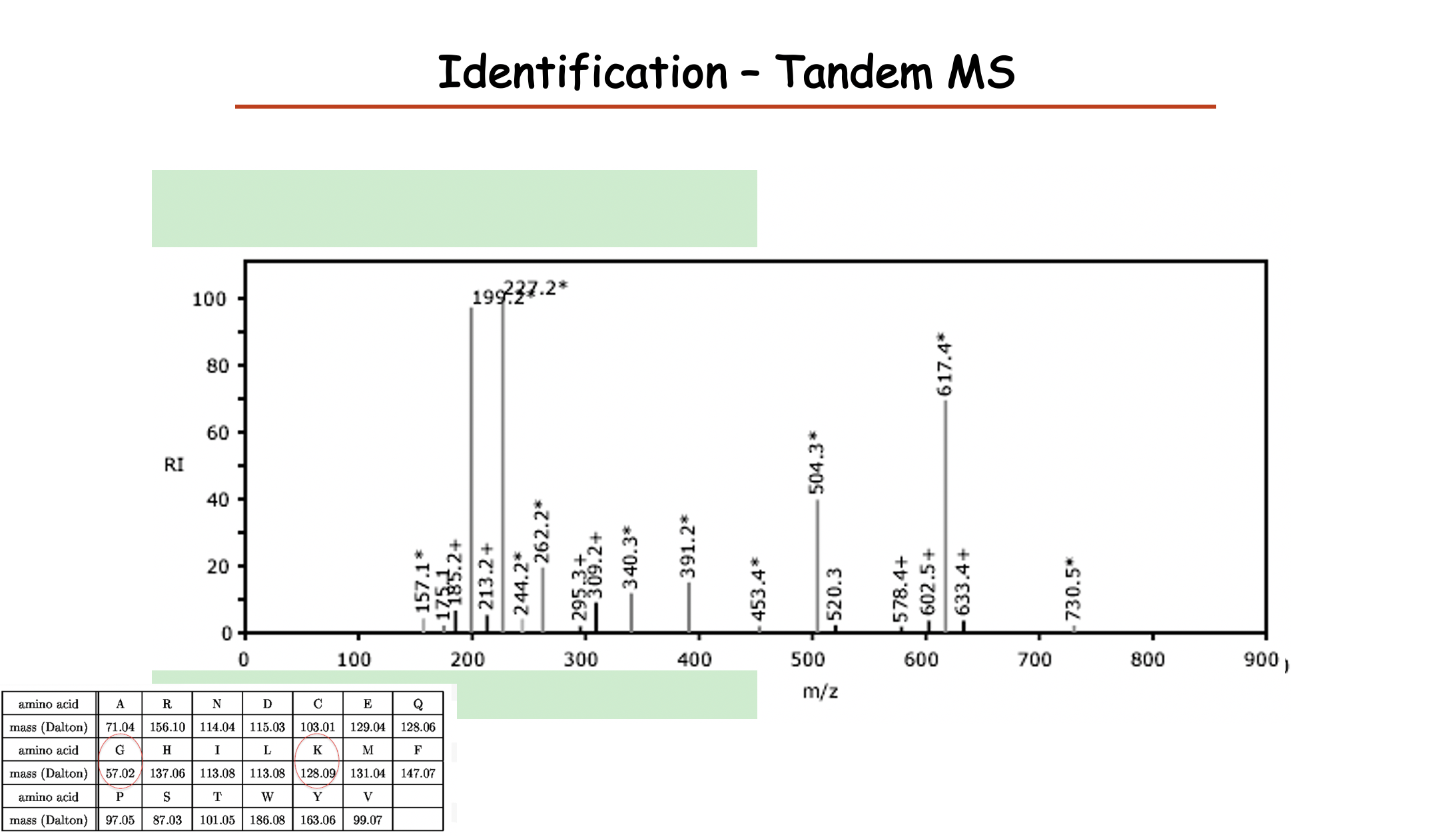
Peak: molecular weight to the ions
为什么会有peak 高低
- Digestion efficiency for some peptide lysing
- Ionize of each protein
可能是问题2的答案
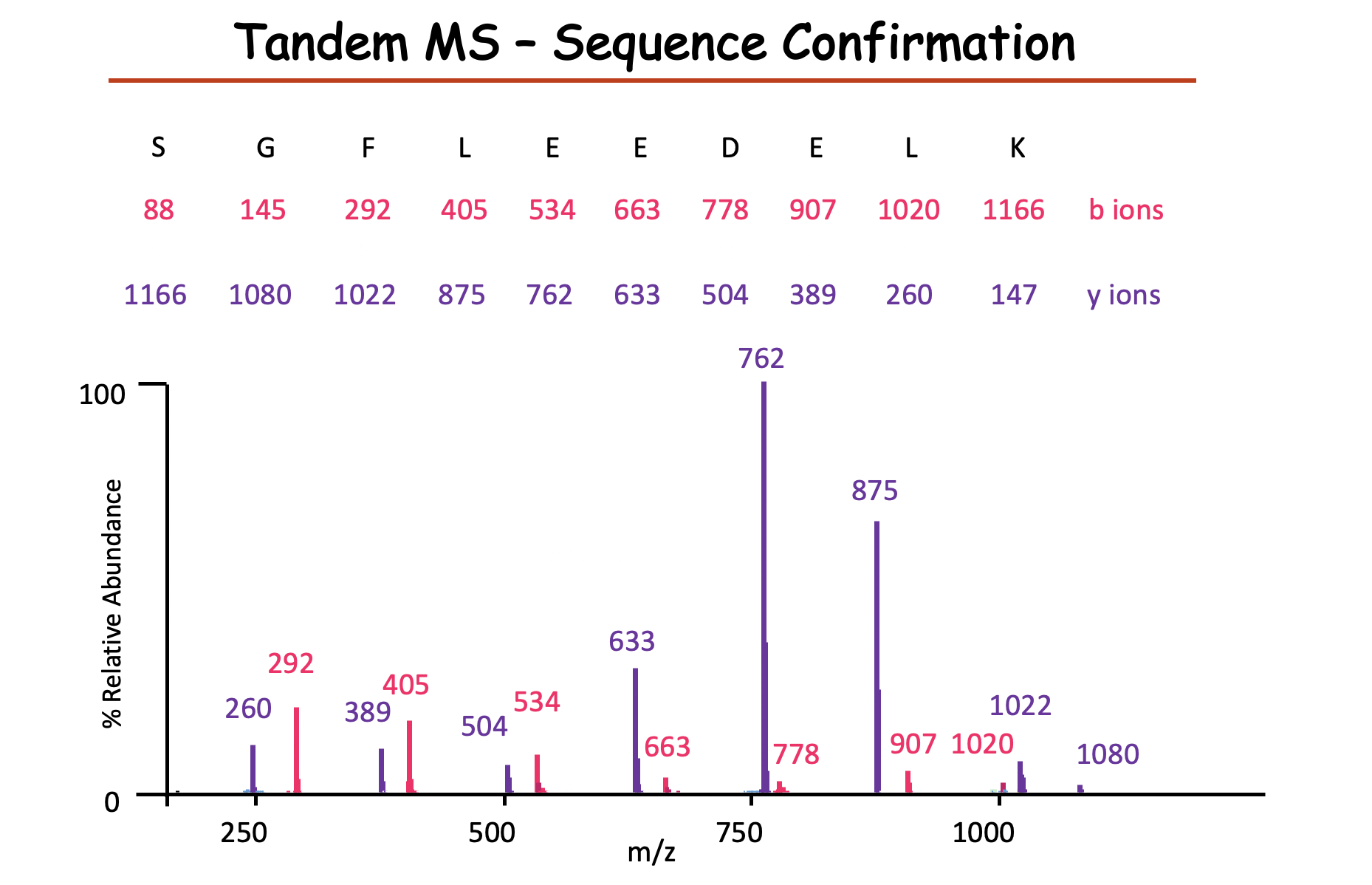
y ions 从右往左数,需要先加上一个18,最后加上1
b ions 从左往右数,需要先加上1,最后加上18
cw2
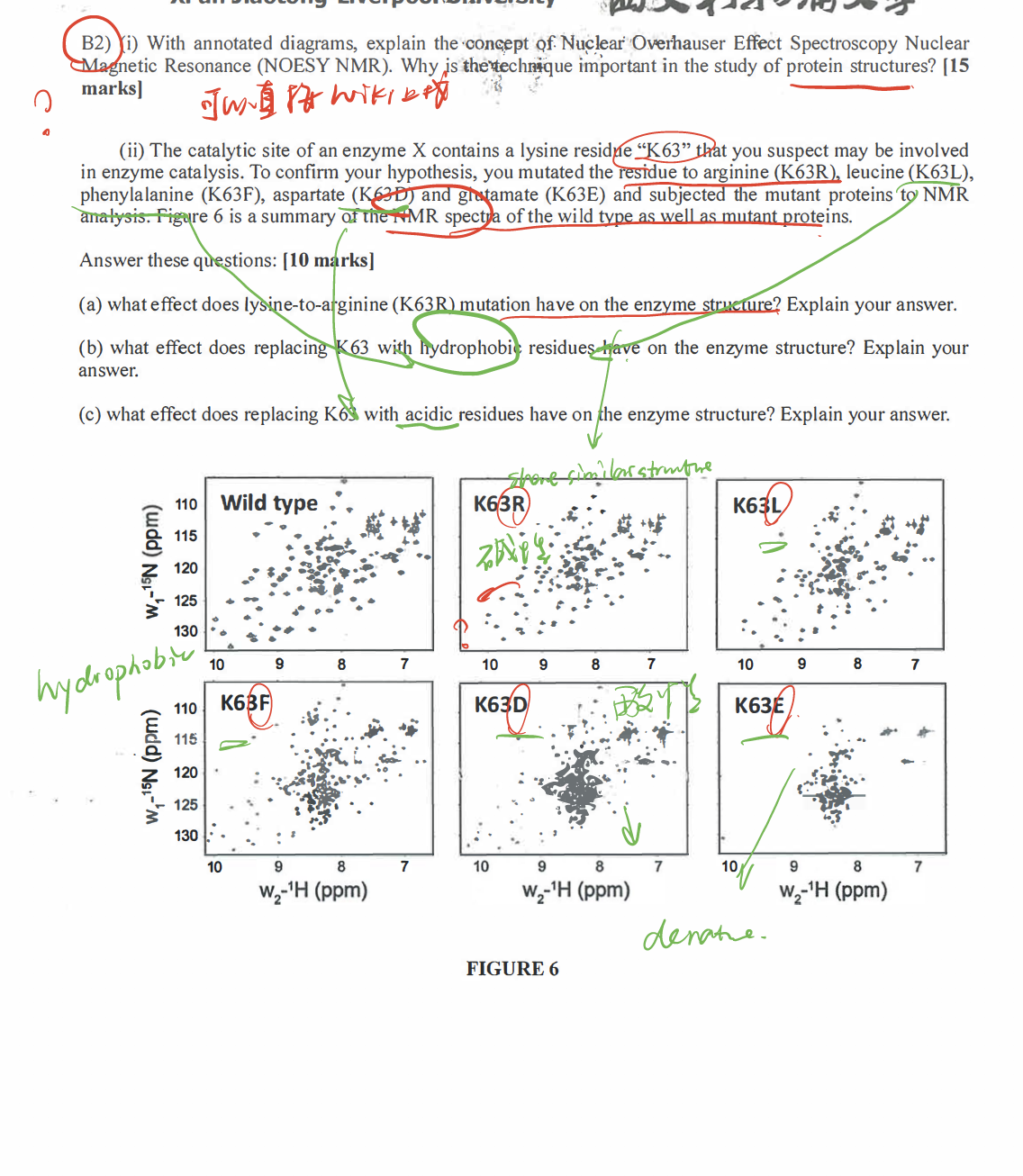
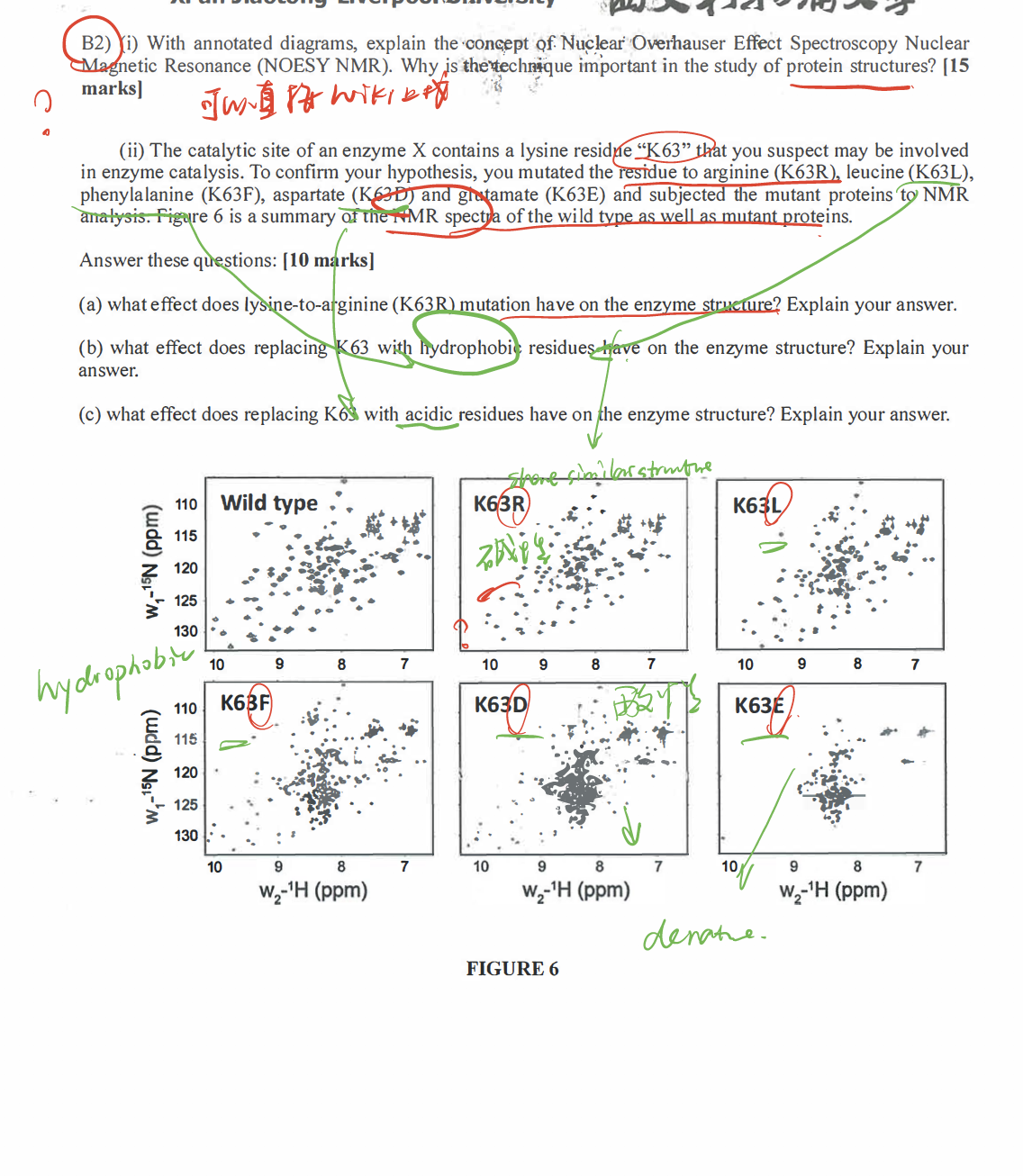
- A redox protein can form many disulfide bonds that are regulatory to its activity. Identification of the disulfide bonds and cysteine residues involved in forming them could be a challenging task.
Mutational analysis(e.g. changing cys to ala) has been the most investigators’ choice. But this method can be very labor-intensive. With the development in ==mass spectrometry==, a mass spec-based disulfide bond determination has become more and more popular. For protein identification using MS, disulfide bonds are normally reduced first and then alkylated (to prevent reforming of disulfide bonds). However, this procedure will not allow for identification of the cysteines involved in disulfide bond formation. Propose a method toidentify the cysteines involved in disulfide bondformation usingmass specas the analytical tool. (20 marks)
- Mass spectrometry
- Identify the cysteines involved in disulfide bond
For example, simple isotope-coded affinity tag (ICAT)-based approaches (33, 91) that pinpoint cysteinyl residues within complex (impure) protein samples are based on splitting a protein sample and labeling half with a light sulfhydryl-reactive ICAT reagent and the other half with a heavy, stable isotope-labeled sulfhydryl-reactive ICAT reagent, mixing, proteolytically digesting, and then analyzing by mass spectrometry. This results in readily identifiable pairs of mass spectral peaks in single-stage mass spectra that are separated by the mass difference between the light and heavy ICAT tags. Mass mapping and, if necessary, analysis of the tagged peptides by tandem mass spectrometry (MS/MS) can reveal the identity of the labeled cysteine residue(s). (More complex ICAT-based schemes for relative quantification are described next.)
Isotope-coded affinity tag (ICAT)-based approaches could be applied to identify the cysteinyl residues involved in disulfide bond formation using mass spec.
The ICAT approach is achieved by splitting a protein sample and labeling half with a light sulfhydryl-reactive ICAT reagent and the other half with a heavy, stable isotope-labeled sulfhydryl-reactive ICAT reagent, mixing, proteolytically digesting, and then analyzing by mass spectrometry.
This results in readily identifiable pairs of mass spectral peaks in single-stage mass spectra that are separated by the mass difference between the light and heavy ICAT tags.
Mass mapping and analysis of the tagged peptides by tandem mass spectrometry (MS/MS) can reveal the identity of the labeled cysteine residue(s).
- You have just joined a biotechnology company and have been told to work on the BSc protein, which is a regulatory protein of therapeutic interest. Specifically, your task is to discover proteins whose cellular levels are influenced by the BSc protein. Describe a mass spec-based proteomic approaches you would use to identify such proteins [20 marks].
- BSc protein (a regulatory protein of therapeutic interest)
- discover proteins whose
cellular levelsare influenced by the BSc protein - mass spec-based proteomic approaches
identify
Answer;
From an MS-based proteomics perspective, all these challenges in cellular levels could be reflected by the quantification of the relative abundance of peptides in different samples. In this case, two technologies including isotope labeling and label-free quantification were used. Consequently, we could compare the abundance between control group and BSc protein influenced group
iTRAQ (isobaric Tags for Relative and Absolute Quantitation), one of the Stable-isotope Labeled Methods, uses up to eight isobaric tags that react with primary amine groups of peptides. During MS analysis, the tags are fragmented into reporter groups of different mass for each tag. The intensity of the different reporter groups is then used to derive the relative abundance of the corresponding peptides and proteins in the mixture.
Besides stable isotope labeling, so-called “label-free” quantification is increasingly used. The basic idea here is to align separate LC-MS/MS runs of peptide mixtures and to calculate differences in intensities of the same peptides detected in each run. Although quantification using this methodology is less accurate than methods using isotope labels, it may be simpler than isotope-based methods and makes cell types accessible that are difficult or impossible to label with amino acids.
The next part would be the data analysis, steps like quality control or normalization might be used for better performance and accurate identification.
From an MS perspective, all these challenges necessitate the quantification of the relative abundance of peptides in different samples. Comparing two protein complexes or large assemblies, such as the nucleolus, requires knowing the relative abundance of peptides derived from proteins present in two preparations, whereas elucidation of signal response requires measuring changes in the levels of phosphopeptides.Several approaches have been developed to tackle this problem.
Mostly, we will focus here on two technologies that are frequently used and that can be combined seamlessly with the analysis pipeline described: isotope labeling and label-free quantification.
In one of the metabolic labeling approaches, proteins fully incorporate “heavy”, nonradioactive isotope-containing amino acids (stable isotope labeling with amino acids in cell culture [SILAC]; Ong et al., 2002) and are analyzed by high resolution MS. To this end, arginine and/or lysine that is labeled with 13C atoms and/or 15N atoms is fed to cells, which integrate these amino acids into all proteins in the course of several cell doublings.
Digestion of these proteins with trypsin, which cuts after arginine or lysine, leads to peptides with a heavy amino acid at their C terminus. The heavy labeled proteome remains distinguishable from the “light” or normal labeled control proteome and the two can be combined at the level of cells or directly after lysis.
This prevents differences in sample preparation from influencing quantification accuracy. The resulting mixture contains SILAC peptide pairs recognizable by having the exact mass difference between the heavy and normal amino acids. The relative intensity of the peaks reflects the relative abundance of the proteins in the mixture.
Although SILAC was originally developed for work on cell lines, it has been extended to include microorganisms and entire mice and even to the very accurate quantification of the levels of thousands of proteins in human tumor biopsies (Krüger et al., 2008; Geiger et al., 2010).
The alternative to metabolic labeling is chemical modification of peptides by stable isotope–containing tags. The best-known strategy to this end is called iTRAQ. It uses up to eight isobaric tags that react with primary amine groups of peptides. During MS analysis, the tags are fragmented into reporter groups of different mass for each tag. The intensity of the different reporter groups is then used to derive the relative abundance of the corresponding peptides and proteins in the starting mixture (Ross et al., 2004).
Besides stable isotope labeling, so-called “label-free” quantification is increasingly used (Old et al., 2005). The basic idea here is to align separate LC-MS/MS runs of peptide mixtures and to calculate differences in intensities of the same peptides detected in each run. Although quantification using this methodology is less accurate than methods using isotope labels, it may be simpler than isotope-based methods and makes cell types accessible that are difficult or impossible to label with amino acids (Malmström et al., 2009; Luber et al., 2010).
In addition to these two, a number of variations and alternative techniques have been developed that are reviewed in detail elsewhere (Ong and Mann, 2005; Bantscheff et al., 2007; Wilm, 2009). As an example, chemical dimethyl labeling of peptides can be performed economically with isotopically light or heavy reagents (Hsu et al., 2003; Boersema et al., 2009). As usual with chemical techniques in proteomics, it is important to ensure that reactions go to completion. Last not least, if quantification of only a selected subset of proteins is desired, peptides of these proteins can be targeted by a technique called multiple reaction monitoring, or MRM. This requires specialized “triple quadrupole” mass spectrometers, which consist of a selection quadrupole for the precursor ion, a collision cell quadrupole, and a selection quadrupole for the fragments. They are set to exclusively monitor predetermined precursor-to-fragment transitions in rapid succession. In this way the presence and—if an isotope-labeled synthetic peptide analogue is used, the quantity of selected peptides—can be monitored
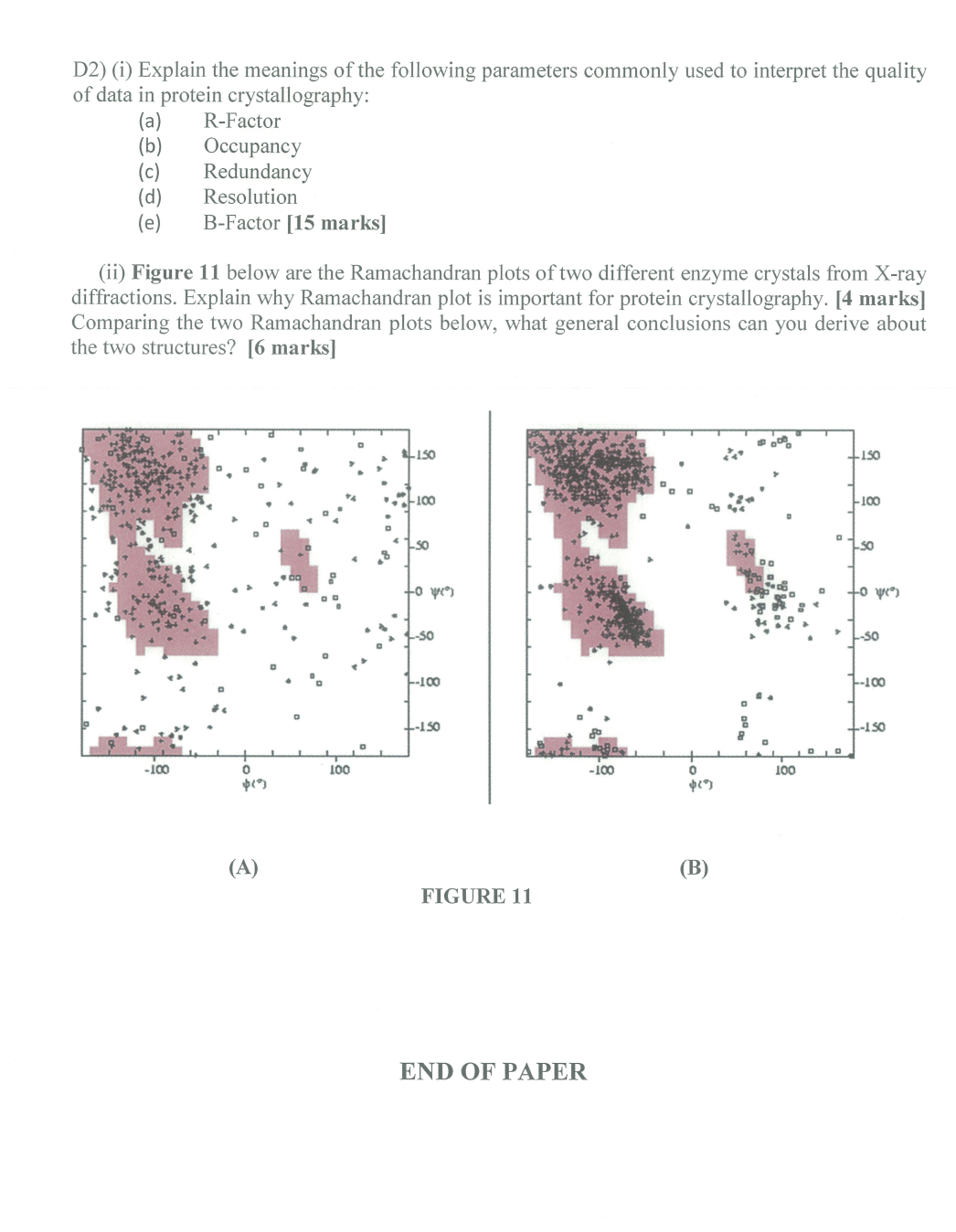
Past exams










- Post title:BIO312 Wang Mu's CW
- Post author:Yuxuan Wu
- Create time:2021-03-10 05:50:03
- Post link:yuxuanwu17.github.io2021/03/10/BIO312 Wang Mu's CW/
- Copyright Notice:All articles in this blog are licensed under BY-NC-SA unless stating additionally.